Elbow angle: Difference between revisions
Jaredsampson (talk | contribs) (→Examples: added more details to first example, and better explanation for Pymol-script-repo example) |
Jaredsampson (talk | contribs) (→Examples: added contents of example script) |
||
| Line 18: | Line 18: | ||
</syntaxhighlight> | </syntaxhighlight> | ||
Note that if you don't specify the light/heavy chains or the variable domain limits, the default (L/H and 107/113 respectively) will be assumed. If your antibody is not Kabat or Chothia numbered, this will result in an incorrect value! Have a look at Dr. Andrew Martin's [http://www.bioinf.org.uk/abs/abnum/ ABNUM] for more information. | Note that if you don't specify the light/heavy chains or the variable domain limits, the default (L/H and 107/113 respectively) will be assumed. If your antibody is not Kabat or Chothia numbered, this will result in an incorrect value! Have a look at Dr. Andrew Martin's [http://www.bioinf.org.uk/abs/abnum/ ABNUM] for more information. | ||
There is also an example .pml file in the Pymol-script-repo/examples directory which can be run by the following: | There is also an example .pml file in the Pymol-script-repo/examples directory which can be run by the following: | ||
| Line 25: | Line 24: | ||
ex elbow_angle | ex elbow_angle | ||
</syntaxhighlight> | </syntaxhighlight> | ||
This will run the following .pml file: | |||
<syntaxhighlight lang="python"> | |||
reinitialize | |||
import elbow_angle | |||
bg_color white | |||
# example structures from Stanfield, et al. JMB 2006 | |||
# doi:10.1016/j.jmb.2006.01.023 | |||
fetch 1bbd, async=0 | |||
fetch 7fab, async=0 | |||
fetch 1dba, async=0 | |||
fetch 1plg, async=0 | |||
fetch 1nl0, async=0 | |||
dss | |||
as cartoon | |||
set cartoon_transparency, 0.7 | |||
remove all and not chain L+H | |||
util.mass_align("1bbd and ((chain L and resi 1-114) or (chain H and resi 1-118))") | |||
# adopt a similar view to Figure 1 in Stanfield, et al. | |||
set_view (\ | |||
-0.953261435, -0.226005003, 0.200535893,\ | |||
-0.230026290, 0.112494141, -0.966659248,\ | |||
0.195909262, -0.967606425, -0.159222543,\ | |||
0.000000000, 0.000000000, -230.122619629,\ | |||
62.279075623, 48.879341125, 138.177505493,\ | |||
181.430419922, 278.814819336, -20.000000000 ) | |||
# 1bbd | |||
# Stanfield: 127 deg | |||
# elbow_angle.py: 125 deg | |||
elbow_angle 1bbd, limit_l=114, limit_h=118, draw=1 | |||
# 7fab | |||
# Stanfield: 132 deg | |||
# elbow_angle.py: 126 deg | |||
elbow_angle 7fab, limit_l=104, limit_h=117, draw=1 | |||
# 1dba | |||
# Stanfield: 183 deg | |||
# elbow_angle.py: 176 deg | |||
elbow_angle 1dba, draw=1 | |||
# 1plg | |||
# Stanfield: 190 deg | |||
# elbow_angle.py: 189 deg | |||
elbow_angle 1plg, limit_l=112, limit_h=117, draw=1 | |||
# 1nl0 | |||
# Stanfield: 220 deg | |||
# elbow_angle.py: 221 deg | |||
elbow_angle 1nl0, draw=1 | |||
</syntaxhighlight> | |||
and will produce the second image below. | |||
<gallery heights="320" widths="320"> | <gallery heights="320" widths="320"> | ||
Revision as of 16:25, 14 June 2012
| Type | Python Script |
|---|---|
| Download | elbow_angle.py |
| Author(s) | Jared Sampson |
| License | GPLv3 |
| This code has been put under version control in the project Pymol-script-repo | |
Introduction
This script allows you to calculate the elbow angle of an antibody Fab fragment object and optionally draw a graphical representation of the vectors used to calculate the elbow angle.
Examples
Basic usage:
# load an antibody Fab fragment from the PDB
fetch 3ghe, async=0
# calculate the elbow angle and draw the vectors
elbow_angle 3ghe, draw=1
Note that if you don't specify the light/heavy chains or the variable domain limits, the default (L/H and 107/113 respectively) will be assumed. If your antibody is not Kabat or Chothia numbered, this will result in an incorrect value! Have a look at Dr. Andrew Martin's ABNUM for more information.
There is also an example .pml file in the Pymol-script-repo/examples directory which can be run by the following:
import ex
ex elbow_angle
This will run the following .pml file:
reinitialize
import elbow_angle
bg_color white
# example structures from Stanfield, et al. JMB 2006
# doi:10.1016/j.jmb.2006.01.023
fetch 1bbd, async=0
fetch 7fab, async=0
fetch 1dba, async=0
fetch 1plg, async=0
fetch 1nl0, async=0
dss
as cartoon
set cartoon_transparency, 0.7
remove all and not chain L+H
util.mass_align("1bbd and ((chain L and resi 1-114) or (chain H and resi 1-118))")
# adopt a similar view to Figure 1 in Stanfield, et al.
set_view (\
-0.953261435, -0.226005003, 0.200535893,\
-0.230026290, 0.112494141, -0.966659248,\
0.195909262, -0.967606425, -0.159222543,\
0.000000000, 0.000000000, -230.122619629,\
62.279075623, 48.879341125, 138.177505493,\
181.430419922, 278.814819336, -20.000000000 )
# 1bbd
# Stanfield: 127 deg
# elbow_angle.py: 125 deg
elbow_angle 1bbd, limit_l=114, limit_h=118, draw=1
# 7fab
# Stanfield: 132 deg
# elbow_angle.py: 126 deg
elbow_angle 7fab, limit_l=104, limit_h=117, draw=1
# 1dba
# Stanfield: 183 deg
# elbow_angle.py: 176 deg
elbow_angle 1dba, draw=1
# 1plg
# Stanfield: 190 deg
# elbow_angle.py: 189 deg
elbow_angle 1plg, limit_l=112, limit_h=117, draw=1
# 1nl0
# Stanfield: 220 deg
# elbow_angle.py: 221 deg
elbow_angle 1nl0, draw=1
and will produce the second image below.
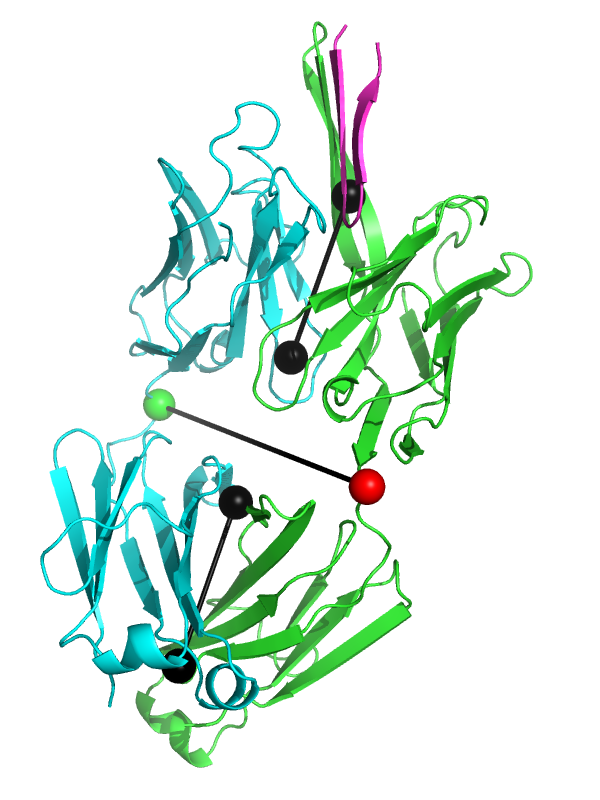
Fab fragment 3ghe shown with draw=1.
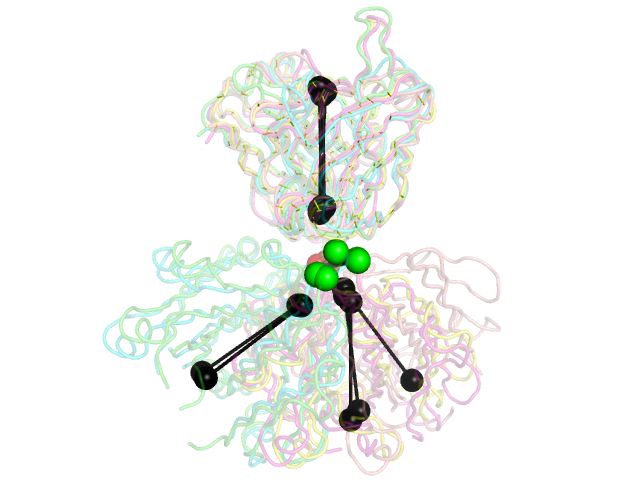
5 PDB examples from Stanfield, et al., JMB 2006, shown in the same orientations as in Figure 1 of that paper.


The black "dumbbells" pass through the centers of mass of the variable and constant domains of each Fab. The green and red dumbbells denote the residues used to split the variable and constant domains, with a green ball for the light chain, and a red ball for the heavy chain.