Elbow angle: Difference between revisions
Jaredsampson (talk | contribs) (→Examples: removed gallery dimensions) |
PedroLacerda (talk | contribs) No edit summary |
||
| (22 intermediate revisions by 2 users not shown) | |||
| Line 1: | Line 1: | ||
{{Infobox script-repo | {{Infobox script-repo | ||
|type = script | |type = script | ||
|filename = elbow_angle.py | |filename = scripts/elbow_angle.py | ||
|author = [[User:jaredsampson|Jared Sampson]] | |author = [[User:jaredsampson|Jared Sampson]] | ||
|license = | |license = BSD | ||
}} | }} | ||
== Introduction == | == Introduction == | ||
This script allows you to calculate the elbow angle of an antibody Fab fragment object and optionally draw a graphical representation of the vectors used to calculate the elbow angle. | This script allows you to calculate the elbow angle of an antibody Fab fragment object and optionally draw a graphical representation of the vectors used to calculate the elbow angle. | ||
== Syntax == | |||
<syntaxhighlight lang="python"> | |||
elbow_angle object [, light=L [, heavy=H [, limit_l=107 [, limit_h=113 [, draw=0 ]]]]] | |||
</syntaxhighlight> | |||
The first argument should be a PyMOL object. You can calculate the elbow angles of multiple Fabs from the same object (one at a time) by specifying the chains manually for each Fab. | |||
The `light` and `heavy` arguments are for PDB chain IDs, and `limit_l` and `limit_h` are the last residue of the light and heavy chain variable domains, respectively. (For Kabat-numbered structures, these limits will be 107 and 113, respectively. For structures with different numbering schemes, the limits can be estimated by visual inspection of the PDB file.) Setting `draw=1` will draw the "dumbbells" seen in the images below. | |||
== Examples == | == Examples == | ||
===Basic usage=== | |||
<syntaxhighlight lang="python"> | <syntaxhighlight lang="python"> | ||
# load an antibody Fab from the PDB | # load an antibody Fab fragment from the PDB | ||
fetch 3ghe, async=0 | fetch 3ghe, async=0 | ||
# calculate the elbow angle and draw the vectors | # calculate the elbow angle and draw the vectors | ||
elbow_angle 3ghe, draw=1 | elbow_angle 3ghe, draw=1 | ||
</syntaxhighlight> | </syntaxhighlight> | ||
This will produce something like the first image below. | |||
<gallery> | Note that if you don't specify the light/heavy chain IDs or the variable domain limit residue numbers, the default values (L/H and 107/113, respectively) will be used. If your antibody is not Kabat or Chothia numbered, or has different chain names, this will result in an incorrect value or, in the case of wrong chain IDs, may cause the script to fail entirely due to an empty selection. Have a look at Dr. Andrew Martin's [http://www.bioinf.org.uk/abs/abnum/ Abnum] for more information on antibody numbering. | ||
<gallery heights="320" widths="320"> | |||
Image:elbow_angle_3ghe.png|Fab fragment 3ghe shown with draw=1. | Image:elbow_angle_3ghe.png|Fab fragment 3ghe shown with draw=1. | ||
Image: | Image:Stanfield.png|5 PDB examples from Stanfield, et al., JMB 2006, shown in the same orientations as in Figure 1 of that paper. | ||
</gallery> | </gallery> | ||
The black "dumbbells" pass through the centers of mass of the | The black "dumbbells" pass through the centers of mass of the variable and constant domains of each Fab (as determined using [[com|com.py]]). The green and red dumbbells denote the residues used to split the variable and constant domains, with a green ball for the light chain, and a red ball for the heavy chain. | ||
===Included Example=== | |||
There is also an example .pml file in the Pymol-script-repo/examples directory which can be run by the following: | |||
<syntaxhighlight lang="python"> | |||
import ex | |||
ex elbow_angle | |||
</syntaxhighlight> | |||
This will run the following .pml file: | |||
{{Infobox script-repo | |||
|type = pml | |||
|filename = examples/elbow_angle.pml | |||
|author = [[User:jaredsampson|Jared Sampson]] | |||
|license = BSD | |||
}} | |||
<include src="https://raw.github.com/Pymol-Scripts/Pymol-script-repo/master/examples/elbow_angle.pml" highlight="python" /> | |||
and will produce the second image above. | |||
==Implementation== | |||
The elbow angle is the angle between the pseudo-twofold axes between the light and heavy chain variable and constant domains, respectively. The rotation matrix to superpose VL onto VH and CL onto CH are calculated and the vectors corresponding to the rotation axes are determined (using Christoph Gohlke's [[transformations|transformations.py]]). The elbow angle is the obtuse angle obtained by taking the arccos of the dot product of the two vectors. For consistency, the elbow angle is reported as less than 180° when the cross product of the Variable and Constant domain rotation axis vectors ( '''V''' ⨯ '''C''' ) is a vector pointing the same direction as that from the limit_h residue alpha carbon atom to the limit_l alpha carbon atom. | |||
[[Category:Script_Library]] | [[Category:Script_Library]] | ||
[[Category:Structural_Biology_Scripts]] | [[Category:Structural_Biology_Scripts]] | ||
[[Category:Pymol-script-repo]] | [[Category:Pymol-script-repo]] | ||
Latest revision as of 21:37, 22 June 2025
| Type | Python Script |
|---|---|
| Download | scripts/elbow_angle.py |
| Author(s) | Jared Sampson |
| License | BSD |
| This code has been put under version control in the project Pymol-script-repo | |
Introduction
This script allows you to calculate the elbow angle of an antibody Fab fragment object and optionally draw a graphical representation of the vectors used to calculate the elbow angle.
Syntax
elbow_angle object [, light=L [, heavy=H [, limit_l=107 [, limit_h=113 [, draw=0 ]]]]]
The first argument should be a PyMOL object. You can calculate the elbow angles of multiple Fabs from the same object (one at a time) by specifying the chains manually for each Fab.
The `light` and `heavy` arguments are for PDB chain IDs, and `limit_l` and `limit_h` are the last residue of the light and heavy chain variable domains, respectively. (For Kabat-numbered structures, these limits will be 107 and 113, respectively. For structures with different numbering schemes, the limits can be estimated by visual inspection of the PDB file.) Setting `draw=1` will draw the "dumbbells" seen in the images below.
Examples
Basic usage
# load an antibody Fab fragment from the PDB
fetch 3ghe, async=0
# calculate the elbow angle and draw the vectors
elbow_angle 3ghe, draw=1
This will produce something like the first image below.
Note that if you don't specify the light/heavy chain IDs or the variable domain limit residue numbers, the default values (L/H and 107/113, respectively) will be used. If your antibody is not Kabat or Chothia numbered, or has different chain names, this will result in an incorrect value or, in the case of wrong chain IDs, may cause the script to fail entirely due to an empty selection. Have a look at Dr. Andrew Martin's Abnum for more information on antibody numbering.
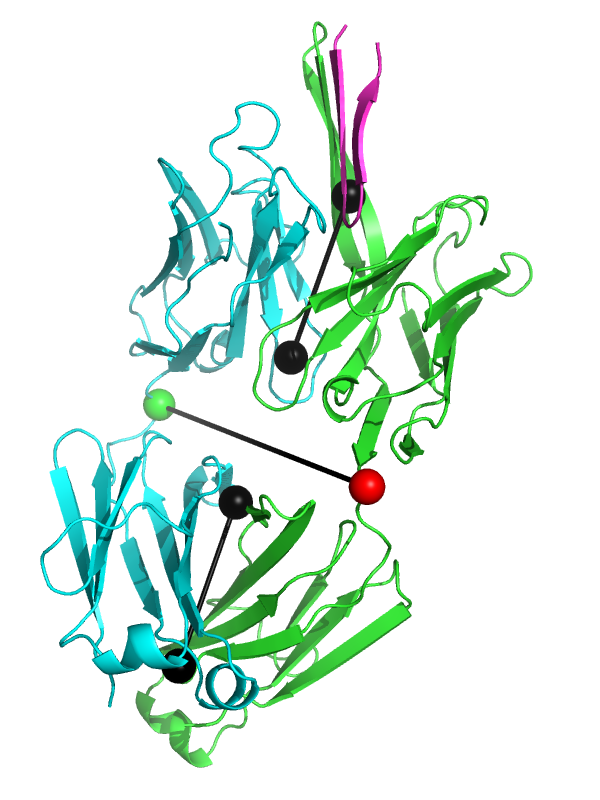
Fab fragment 3ghe shown with draw=1.
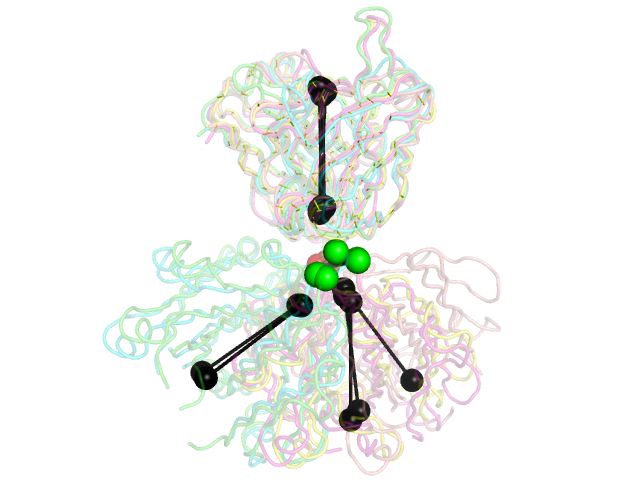
5 PDB examples from Stanfield, et al., JMB 2006, shown in the same orientations as in Figure 1 of that paper.


The black "dumbbells" pass through the centers of mass of the variable and constant domains of each Fab (as determined using com.py). The green and red dumbbells denote the residues used to split the variable and constant domains, with a green ball for the light chain, and a red ball for the heavy chain.
Included Example
There is also an example .pml file in the Pymol-script-repo/examples directory which can be run by the following:
import ex
ex elbow_angle
This will run the following .pml file:
| Type | PyMOL Script |
|---|---|
| Download | examples/elbow_angle.pml |
| Author(s) | Jared Sampson |
| License | BSD |
| This code has been put under version control in the project Pymol-script-repo | |
<include src="https://raw.github.com/Pymol-Scripts/Pymol-script-repo/master/examples/elbow_angle.pml" highlight="python" />
and will produce the second image above.
Implementation
The elbow angle is the angle between the pseudo-twofold axes between the light and heavy chain variable and constant domains, respectively. The rotation matrix to superpose VL onto VH and CL onto CH are calculated and the vectors corresponding to the rotation axes are determined (using Christoph Gohlke's transformations.py). The elbow angle is the obtuse angle obtained by taking the arccos of the dot product of the two vectors. For consistency, the elbow angle is reported as less than 180° when the cross product of the Variable and Constant domain rotation axis vectors ( V ⨯ C ) is a vector pointing the same direction as that from the limit_h residue alpha carbon atom to the limit_l alpha carbon atom.