LigAlign
IMAGES
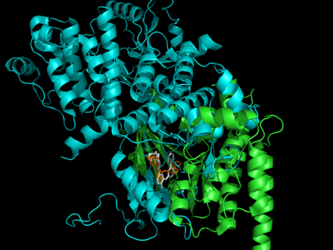
Ligand-based alignment of 1XBB and 1OPJ
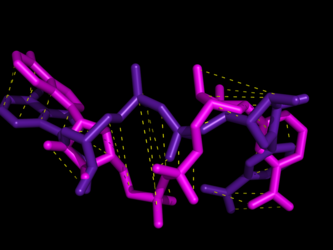
Atom-to-atom mapping of ligands
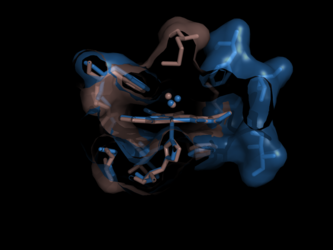
Alignment of 1V07 and 1HBI using heme



DESCRIPTION
LigAlign is a tool to compare protein active-sites and investigate ligand binding. The active-site alignment is guided by the orientation of bound ligands in the protein active sites. LigAlign supports analysis of flexible ligand via automatic fragment-based alignment: first computing a natural fragmentation of the query ligand, aligning each fragment of the query independently against the baseline, and then permitting easy visualization of each active site subcavity.
We use protein-ligand complexes to compare the active sites of several proteins which interact with a chosen ligand. Beginning with a user-specified protein-ligand structure, LigAlign gathers experimental structures of other proteins bound to the ligand from the Protein Data Bank. The tool then aligns the ligands bound in the structures to minimize the ligand-to-ligand RMSD. This transformation also aligns the active sites. Finally, the user can examine the aligned active sites to identify structural patterns, such as conserved steric hindrance or hydrophobicity.
However, a flexible ligand can bend itself into different active sites, where the active site subcavities have different relative positions or orientations. Therefore, when comparing two active sites, a rigid RMSD-minimizing transform on docked flexible ligands may fail to align the correct portions of the active site. However, each subcavity should still exhibit chemical or geometric complementarity to the piece of the ligand which it binds. Please see the website for more information.
LigAlign simplifies a number of protein analysis tasks. For example, LigAlign will align similar but distinct ligands which, in the context of structure-based drug discovery, permits the comparison of the docking of different ligands. Alternatively, if the user only specifies one protein-ligand complex, LigAlign will find chemically similar ligands automatically via the Protein-Small Molecule Database. Finally, LigAlign improves workflow by automatically fetching necessary data from the Protein Data Bank.
LigAlign is contributed by Abraham Heifets and Ryan Lilien at the University of Toronto.
Additional Information
See Also
- mcsalign (psico)