FindSurfaceResidues
Jump to navigation
Jump to search
| Type | Python Module |
|---|---|
| Download | scripts/findSurfaceResidues.py |
| Author(s) | Jason Vertrees |
| License | BSD-2-Clause |
| This code has been put under version control in the project Pymol-script-repo | |
The findSurfaceResidues script will select (and color if requested) surface residues and atoms on an object or selection. See the options below.
Each time, the script will create two new selections called, exposed_res_XYZ and exposed_atm_XYZ where XYZ is some random number. This is done so that no other selections/objects are overwritten.
Usage
findSurfaceResidues [ selection=all [, cutoff=2.5 [, doShow=0 ]]]
Arguments
- selection = str: The object or selection for which to find exposed residues {default: all}
- cutoff = float: The cutoff in square Angstroms that defines exposed or not. Those atoms with > cutoff Ang^2 exposed will be considered exposed {default: 2.5 Ang^2}
- doShow = 0/1: Change the visualization to highlight the exposed residues vs interior {default: 0}
Examples
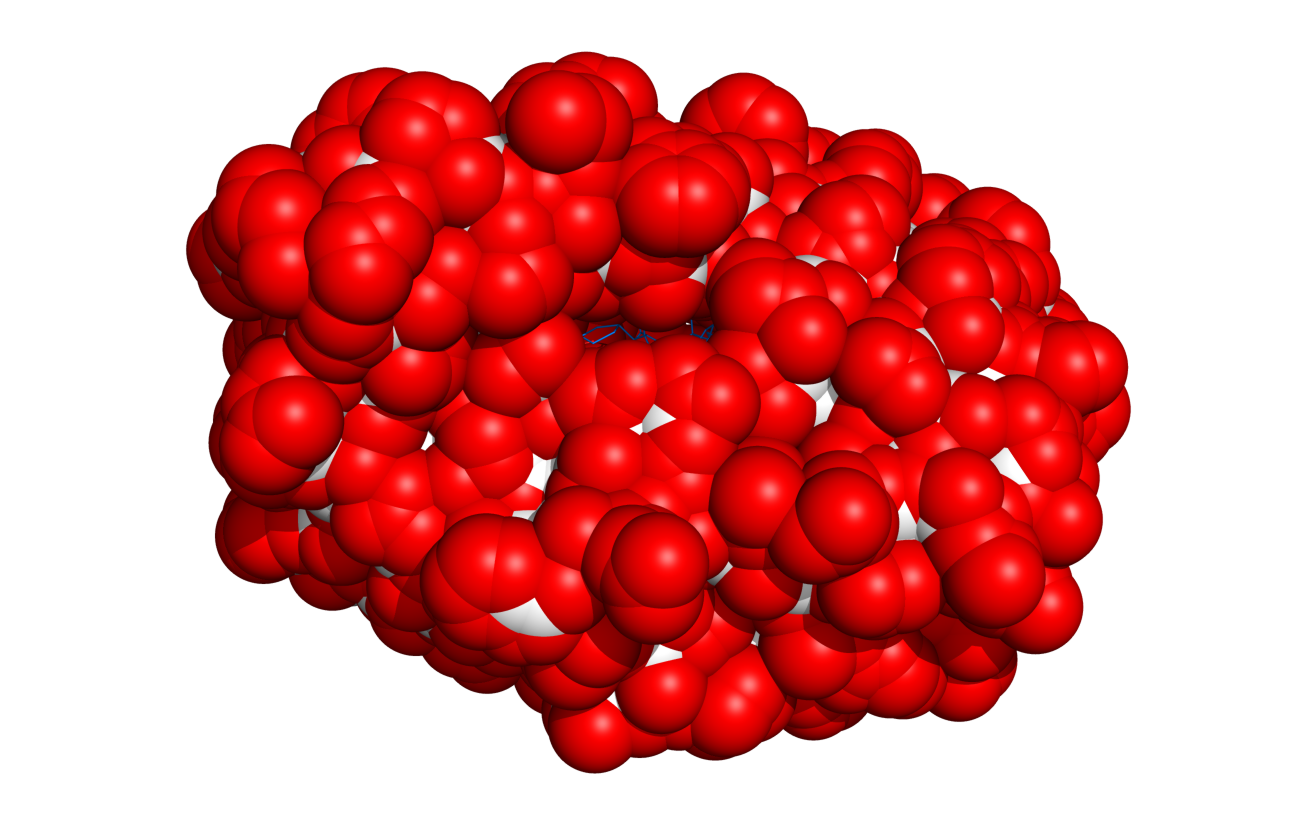
Result of $TUT/1hpv.pdb at 2.5 Ang cutoff.
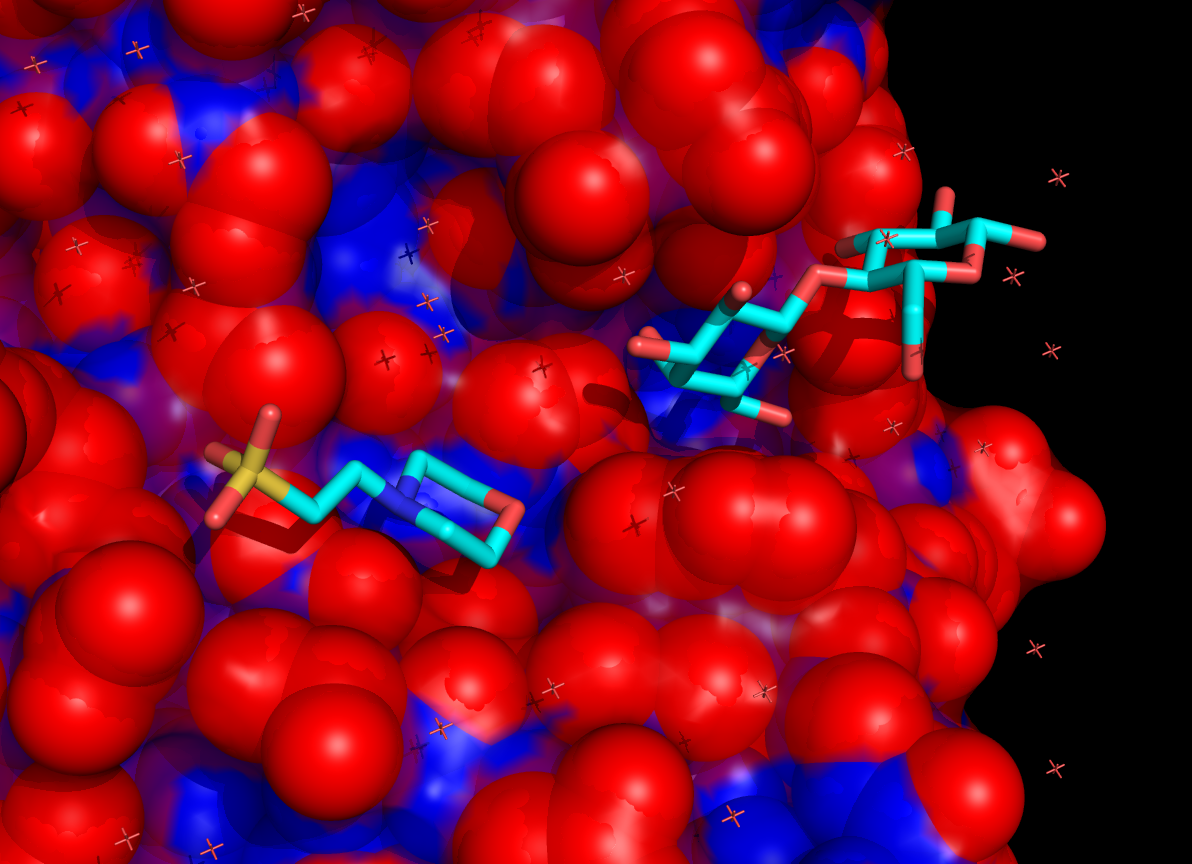
Example coloring of surface residues


run findSurfaceResidues.py
fetch 1hpv, async=0
findSurfaceResidues
# now show the exposed
findSurfaceResidues doShow=1
# watch how the visualization changes:
findSurfaceResidues doShow=1, cutoff=0.5
findSurfaceResidues doShow=1, cutoff=1.0
findSurfaceResidues doShow=1, cutoff=1.5
findSurfaceResidues doShow=1, cutoff=2.0
findSurfaceResidues doShow=1, cutoff=2.5
findSurfaceResidues doShow=1, cutoff=3.0