Difference between revisions of "Draw Protein Dimensions"
| Line 39: | Line 39: | ||
# calculate the dimensions of the full ASU | # calculate the dimensions of the full ASU | ||
draw_Protein_Dimensions 2vak | draw_Protein_Dimensions 2vak | ||
| + | </source> | ||
<gallery perrow=3 widths=350 heights=350> | <gallery perrow=3 widths=350 heights=350> | ||
| − | File:2vak_ASU.png | | + | File:2vak_ASU.png |Dimensions of 2vak ASU |
| + | </gallery> | ||
| + | <source lang="python"> | ||
# you can extract only one chain and calculates the dimensions | # you can extract only one chain and calculates the dimensions | ||
draw_Protein_Dimensions obj01 | draw_Protein_Dimensions obj01 | ||
| + | </source> | ||
| − | + | <gallery perrow=3 widths=350 heights=350> | |
| + | File:2vak_A.png |Dimensions of one protomer (chain A - 2vak) | ||
| + | </gallery> | ||
| + | <source lang="python"> | ||
# You can also draw the Bounding boxes (AABB and IABB) to compare them. | # You can also draw the Bounding boxes (AABB and IABB) to compare them. | ||
| − | |||
fetch 2vak | fetch 2vak | ||
| + | draw_BB 2vak | ||
| + | </source> | ||
| − | |||
| − | |||
| − | |||
| + | <gallery perrow=3 widths=350 heights=350> | ||
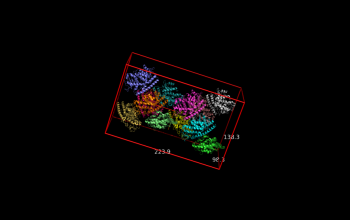
| + | File:2vak_AABB.png |Axis-aligned bounding box | ||
| + | File:2vak_IABB.png |Inertia-axis-aligned bounding box | ||
| + | </gallery> | ||
Latest revision as of 23:52, 2 January 2016
| Type | Python Script |
|---|---|
| Download | Draw_Protein_Dimensions.py |
| Author(s) | Pablo Guardado Calvo |
| License | |
| This code has been put under version control in the project Pymol-script-repo | |
This script will draw the dimensions of a protein based on an Inertia Axis Aligned Bounding Box (IABB).
The idea behind this script is to calculate an approximate minimal bounding box (MBB) to extract the cell dimensions of a protein. To calculate the MBB is not trivial and usually the Axis Aligned Bounding Box (AABB) does not show up the real dimensions of the protein. This script calculates the inertia tensor of the object, extract the eigenvectors and use them to rotate the molecule (using as rotation matrix the transpose of the eigenvector matrix). The result is a molecule oriented with the inertia axis aligned with the cartesian axis. A new Bounding Box is calculated, which is called Inertia Axis Aligned Bounding Box (IABB), with a volume always lower than the AABB volume, and in many cases may correspond with the MBB.
As always with these type of things, you have to use at your own risk. I did not try all the possible combinations, but if you find a bug, do not hesitate to contact me (pablo.guardado (at) gmail.com) or try to modify the code for yourself to correct it.
To load the script just type:
run path-to-the-script/Draw_Protein_Dimensions.py
or if you want something more permanent add the previous line to your .pymolrc file
The script works just typing:
draw_Protein_Dimensions selection
This will draw the cell dimensions of your selection based on a IABB box. It also generates the IABB box and the inertia axis, you just need to do "show cgo" to display them.
You could also try:
draw_BB selection
This will draw the AABB and IABB boxes with their cell dimensions and show up their volumes, you can compare them.
Examples
# download the source and save as Draw_Protein_Dimensions.py
run Draw_Protein_Dimensions.py
fetch 2vak
# calculate the dimensions of the full ASU
draw_Protein_Dimensions 2vak
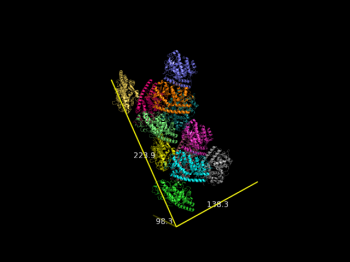
Dimensions of 2vak ASU

# you can extract only one chain and calculates the dimensions
draw_Protein_Dimensions obj01
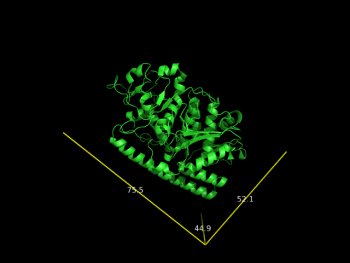
Dimensions of one protomer (chain A - 2vak)

# You can also draw the Bounding boxes (AABB and IABB) to compare them.
fetch 2vak
draw_BB 2vak

Axis-aligned bounding box
Inertia-axis-aligned bounding box

