Format bonds: Difference between revisions
Jump to navigation
Jump to search
(created script page; linked in valence page) |
PedroLacerda (talk | contribs) No edit summary |
||
| Line 1: | Line 1: | ||
{{Infobox script-repo | {{Infobox script-repo | ||
|type = Python Module | |type = Python Module | ||
|filename = format_bonds.py | |filename = scripts/format_bonds.py | ||
|author = [[User:Andwar|Andreas Warnecke]] | |author = [[User:Andwar|Andreas Warnecke]] | ||
|license = BSD-2-Clause | |license = BSD-2-Clause | ||
| Line 42: | Line 42: | ||
[[Category:Script_Library]] | [[Category:Script_Library]] | ||
[[Category:UI_Scripts]] | [[Category:UI_Scripts]] | ||
[[Category:Pymol-script-repo]] | |||
Latest revision as of 23:54, 22 June 2025
| Type | Python Module |
|---|---|
| Download | scripts/format_bonds.py |
| Author(s) | Andreas Warnecke |
| License | BSD-2-Clause |
| This code has been put under version control in the project Pymol-script-repo | |
The script format_bonds will automatically format bonds in amino acids.
Usage
format_bonds [ selection [, bonds ]]
Examples
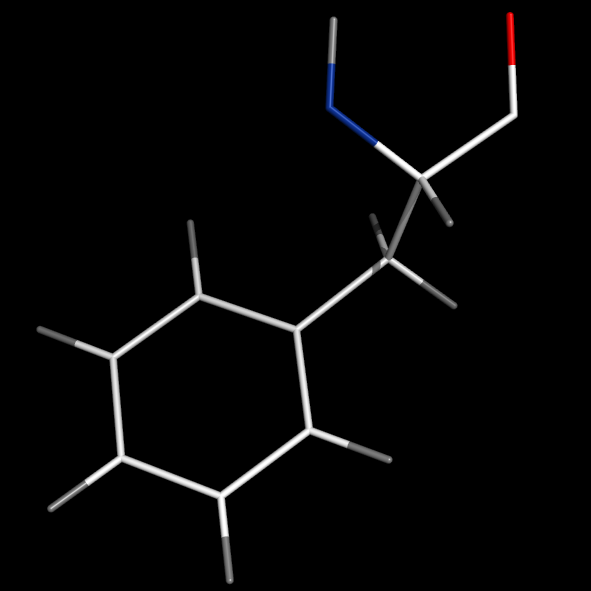
format_bonds bonds=1
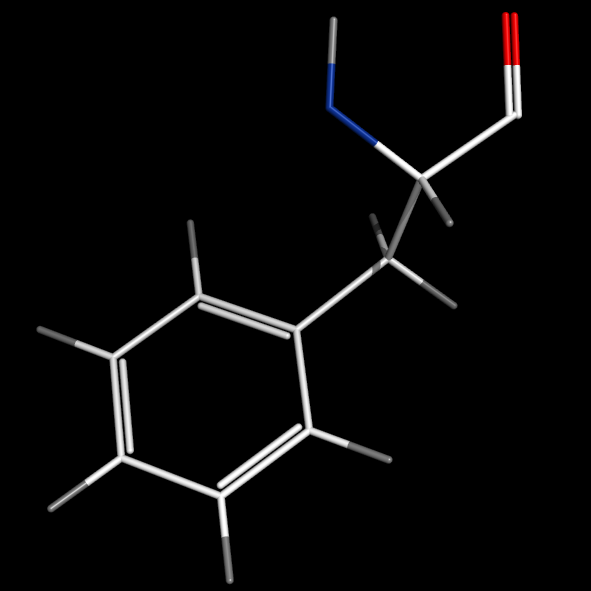
format_bonds bonds=2
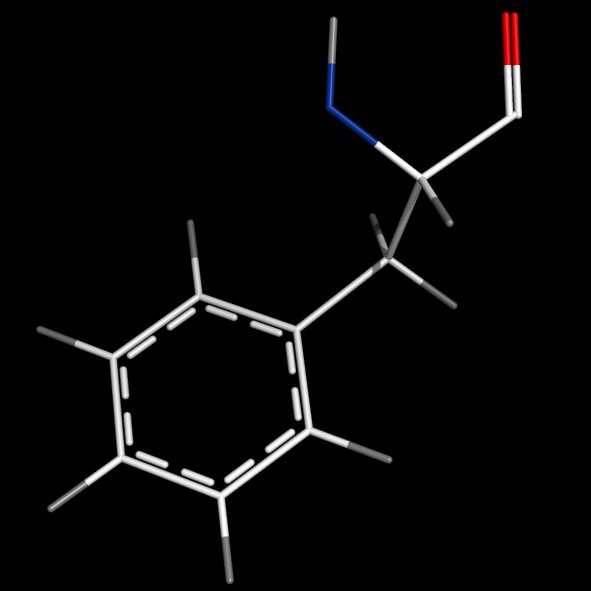
format_bonds



import format_bonds
frag PHE
format_bonds
format_bonds bonds=2
Notes
- Remember to correctly configure plugin import (see: Git intro)
- format_bonds will introduce delocalized bonds by default or when bonds is larger than 2.
- Setting bonds=1 will simply disable valence display (globally!)
- The selection argument is 'all' by default and can be used to restrict editing to selected residues.
- Note that format_bonds will also format acidic residues, the C-terminus, arginine and nitro groups.
- Tip: press the TAB key after entering format_bonds to get argument suggestions