ColorByRMSD: Difference between revisions
No edit summary |
|||
| Line 104: | Line 104: | ||
=== Jason's Attempt === | === Jason's Attempt === | ||
I decided to generalize what Shiven started into an actual function. This code is based off Shiven's (above). | I decided to generalize what Shiven started into an actual function. This code is based off Shiven's (above). Something is still slightly amiss, here: it doesn't work 100% of the time as expected. | ||
==== Examples ==== | ==== Examples ==== | ||
<source lang="python"> | <source lang="python"> | ||
Revision as of 08:02, 16 July 2009
Introduction
An attempt to perform a coloring of two structures by RMS deviation as calculated by PyMol's internal Rms_Cur command.
Code
Shiven's Code
WARNING: This is still a work in progress, and is almost useless right now! It is just a proof of principle at this stage and mainly an attempt at using the "alignment" object to iterate over the aligned objects. However, if you make any improvements, please do edit this page to reflect the same.
"""
--- ColorByRMSD: RMSD based coloring ---
Author : Shivender Shandilya
Program : ColByRMS
Date : July 2009
Version : 0.0.2 (very very alpha!)
Mail : firstname.lastname@umassmed.edu
Keywords: color rms rmsd colorbyrms colorbyrmsd
----------------------------------------------------------------------
Reference:
This email from Warren - http://www.mail-archive.com/pymol-users@lists.sourceforge.net/msg07078.html
Literature:
DeLano, W.L. The PyMOL Molecular Graphics System (2002) DeLano Scientific, San Carlos, CA, USA. http://www.pymol.org
----------------------------------------------------------------------
"""
import pymol
import cmd
# Code for a special case first
cmd.load("D:/PyMOL-1.2r1/data/tut/1hpv.pdb", "1hpv")
cmd.create("chA", "1hpv and polymer and chain A")
cmd.create("chB", "1hpv and polymer and chain B")
# We need the alignment object, but PyMol refuses to give us that unless
# we (needlessly) specify all the other values in the cmd.align() function
# all the 0's are needlessly forced to be specified...
# PyMol is not smart enough to use the defaults
cmd.align("chA and name CA", "chB and name CA",0,0,0,0,0,object="aln")
# So, lets use "super" instead
#cmd.super("chA", "chB",object="aln")
# Utter the magic word
cmd.refresh()
# Arrays to store all residue identifiers in "aln"
stored.alnAres = []
stored.alnBres = []
# Now interrogate "aln" to get residue numbers for each object
cmd.iterate("chA and aln","stored.alnAres.append(resi)")
cmd.iterate("chB and aln","stored.alnBres.append(resi)")
print "Length of alnAres: "+str(len(stored.alnAres))
print "Length of alnBres: "+str(len(stored.alnBres))
# The main function that assigns "cur_rms" as the new b-factor
def colbyRMS(objA, alnAri, objB, alnBri):
cmd.alter(str(objA), "chain='Z'")
cmd.alter(str(objB), "chain='Z'")
cmd.rebuild()
for x in range(len(alnAri)):
rmsd = cmd.rms_cur(str(objA)+" and i. "+str(alnAri[x]), str(objB)+" and i. "+str(alnBri[x]), matchmaker=4)
cmd.alter(str(objA)+" and resi "+str(alnAri[x]), "b = "+str(rmsd))
cmd.alter(str(objB)+" and resi "+str(alnBri[x]), "b = "+str(rmsd))
cmd.extend("colbyRMS", colbyRMS)
# Run the just defined function
if len(stored.alnAres) > len(stored.alnBres):
colbyRMS("chA",stored.alnAres,"chB",stored.alnBres)
else:
colbyRMS("chB",stored.alnBres,"chA",stored.alnAres)
# Arrays to store the NEW b-factors
stored.alnAnb = []
stored.alnBnb = []
# Store the NEW b-factors
cmd.iterate("chA and aln","stored.alnAnb.append(b)")
cmd.iterate("chB and aln","stored.alnBnb.append(b)")
# Assign the just stored NEW b-factors to the original object
for x in range(len(stored.alnAres)):
cmd.alter("1hpv and chain A and resi "+str(stored.alnAres[x]), "b = "+str(stored.alnAnb[x]))
for x in range(len(stored.alnBres)):
cmd.alter("1hpv and chain B and resi "+str(stored.alnBres[x]), "b = "+str(stored.alnBnb[x]))
# Get rid of all intermediate objects etc.
cmd.delete("chA")
cmd.delete("chB")
cmd.delete("aln")
# Showcase what you did
cmd.orient()
cmd.hide("all")
cmd.show("cartoon", "1hpv")
cmd.cartoon("loop")
print "\n"
print "Colored by 'overall' RMSD...\n"
cmd.spectrum("b", "rainbow", "1hpv and polymer")
Jason's Attempt
I decided to generalize what Shiven started into an actual function. This code is based off Shiven's (above). Something is still slightly amiss, here: it doesn't work 100% of the time as expected.
Examples
# example #1
colorByRMSD 1cbs, 1hmt, doAlign=True, doPretty=T
# example #2
colorByRMSD 1eaz, 1fao, doAlign=True, doPretty=T
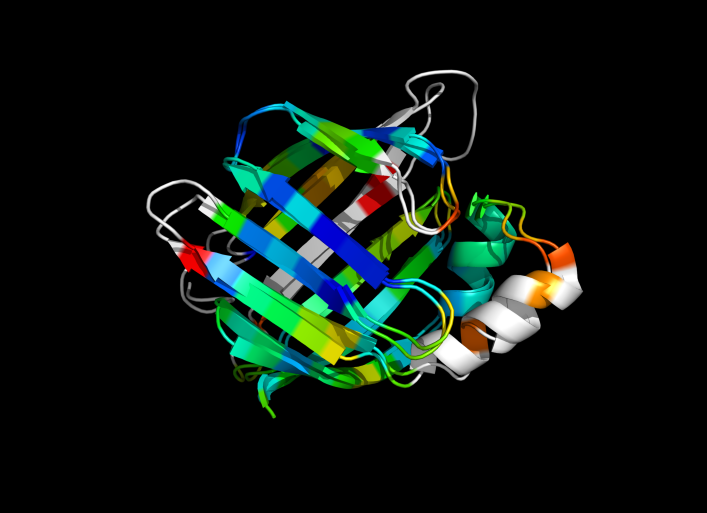
1cbs and 1hmt aligned and colored by RMSD. Dark blue is good alignment, higher deviations are in orange/yellow/red.
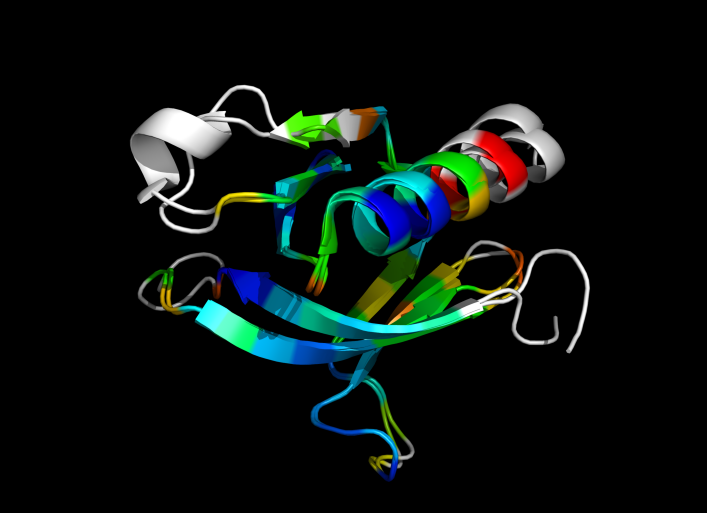
1eaz and 1fao aligned and colored by RMSD. Dark blue is good alignment, higher deviations are in orange/yellow/red.


"""
--- ColorByRMSD: RMSD based coloring ---
Author : Shivender Shandilya, modified by Jason Vertrees
Program : ColByRMS
Date : July 2009
Version : 0.0.2 (very very alpha!)
Mail : firstname.lastname@umassmed.edu
Keywords: color rms rmsd colorbyrms colorbyrmsd
----------------------------------------------------------------------
Reference:
This email from Warren - http://www.mail-archive.com/pymol-users@lists.sourceforge.net/msg07078.html
Literature:
DeLano, W.L. The PyMOL Molecular Graphics System (2002) DeLano Scientific, San Carlos, CA, USA. http://www.pymol.org
----------------------------------------------------------------------
"""
import pymol
import cmd
from pymol import stored
def strTrue(p):
return p[0].upper() == "T"
# The main function that assigns "cur_rms" as the new b-factor
def rmsUpdateB(objA, alnAri, objB, alnBri):
# don't need the *10 -- PyMOL scales things for us.
for x in range(len(alnAri)):
s1 = objA + " and n. CA and i. " + alnAri[x]
s2 = objB + " and n. CA and i. " + alnBri[x]
rmsd = cmd.rms_cur(s1, s2, matchmaker=4)
cmd.alter( s1, "b = " + str(rmsd))
cmd.alter( s2, "b = " + str(rmsd))
cmd.sort(objA); cmd.sort(objB)
def colorByRMSD(objSel1, objSel2, doAlign="True", doPretty=None):
"""
colorByRMSD -- align two structures and show the structural deviations in
color to more easily see variable regions.
PARAMS
objSel1 (valid PyMOL object or selection)
The first object to align.
objSel2 (valid PyMOL object or selection)
The second object to align
doAlign (boolean, either True or False)
Should this script align your proteins or just leave them where they
are? If doAlign=True then your original proteins are aligned. If
doAlign=False, then they are not. Regardless, the b-factors are
changed.
DEFAULT: True
doPretty (boolean, either True or False)
If doPretty=True then a simple representation is created to high-
light the differences. If False, then no change is done to the
structure.
RETURNS
None.
SIDE-EFFECTS
Modified the b-factor columns in your original proteins.
"""
# create backup copies; names starting with _ (underscores) are
# hidden by PyMOL
tObj1, tObj2, aln = "__tempObj1", "__tempObj2", "__aln"
if strTrue(doAlign):
# perform the alignment
cmd.create( tObj1, objSel1 )
cmd.create( tObj2, objSel2 )
cmd.super( tObj1, tObj2, object=aln )
# bug -- every other call undoes this...
cmd.matrix_copy(tObj1, objSel1)
else:
# perform the alignment
cmd.create( tObj1, objSel1 )
cmd.create( tObj2, objSel2 )
cmd.super( tObj1, tObj2, object=aln )
cmd.alter( tObj1 + " or " + tObj2, "b=-10")
cmd.alter( tObj1 + " or " + tObj2, "chain='A'")
cmd.alter( tObj1 + " or " + tObj2, "segi='A'")
# update PyMOL;
# one of these should do the trick
cmd.refresh(); cmd.rebuild(); cmd.sort(tObj1); cmd.sort(tObj2)
# Get the residue identifiers from the aln object
stored.alnAres, stored.alnBres = [], []
cmd.iterate(tObj1 + " and n. CA and " + aln, "stored.alnAres.append(resi)")
cmd.iterate(tObj2 + " and n. CA and " + aln, "stored.alnBres.append(resi)")
# reset the b-factors for each object
rmsUpdateB(tObj1,stored.alnAres,tObj2,stored.alnBres)
# Store the NEW b-factors
stored.alnAnb, stored.alnBnb = [], []
cmd.iterate(tObj1 + " and n. CA and " + aln, "stored.alnAnb.append(b)" )
cmd.iterate(tObj2 + " and n. CA and " + aln, "stored.alnBnb.append(b)" )
# Get rid of all intermediate objects etc.; clean up
cmd.delete(tObj1)
cmd.delete(tObj2)
cmd.delete(aln)
# Assign the just stored NEW b-factors to the original object
for x in range(len(stored.alnAres)):
cmd.alter(objSel1 + " and n. CA and i. " + str(stored.alnAres[x]), "b = " + str(stored.alnAnb[x]))
for x in range(len(stored.alnBres)):
cmd.alter(objSel2 + " and n. CA and i. " + str(stored.alnBres[x]), "b = " + str(stored.alnBnb[x]))
cmd.rebuild(); cmd.refresh(); cmd.sort(objSel1); cmd.sort(objSel2)
if doPretty!=None:
# Showcase what we did
cmd.orient()
cmd.hide("all")
cmd.show_as("cartoon", objSel1 + " or " + objSel2)
cmd.spectrum("b", 'rainbow', "(" + objSel1 + " and n. CA) or (n. CA and " + objSel2 +" )")
cmd.extend("colorByRMSD", colorByRMSD)