Cealign plugin: Difference between revisions
| Line 56: | Line 56: | ||
Image:ali_all.png|All proteins aligned to 1A15 | Image:ali_all.png|All proteins aligned to 1A15 | ||
</gallery> | </gallery> | ||
Paste the following code into the end of the '''cealign.py''' file that comes in the distribution. This function will define the '''alignto''' command in PyMOL. The function will align every object in PyMOL to the specified object. | |||
<source lang="python"> | |||
def alignto(sel1): | |||
"""Just a quick & dirty multiple structure alignment""" | |||
for x in cmd.get_names("*"): cealign( sel1, x ) | |||
## Let PyMOL know about the alignto command | |||
cmd.extend("alignto", alignto) | |||
</source> | |||
Once the code is in place, and PyMOL (or the cealign.py script) is reloaded, you can now execute, | |||
<source lang="python"> | |||
alignto X | |||
</source> | |||
will align all objects in PyMOL to protein '''X'''. | |||
For example, | |||
<source lang="python"> | |||
# for showing pretty representation | |||
import preset | |||
# get a bunch of similar structures and | |||
# load them all at once. | |||
fetch 1a2p 1bni 2f4y 2f56 2f5m 2f5w, async=0 | |||
# poor-man's multiple structure alignment | |||
alignto 1bni | |||
# make them 'pretty'. | |||
preset.pretty("*") | |||
# center the results (since they may all have | |||
# moved out of frame). | |||
center | |||
</source> | |||
=== Results From v0.2 === | === Results From v0.2 === | ||
Revision as of 19:28, 7 March 2007
Introduction
This script is a Python implementation of the CE algorithm pioneered by Drs. Shindyalov and Bourne (See References). It is a fast, accurate structure-based protein alignment algorithm. There are a few changes from the original code (See Notes), and "fast" depends on your machine and the implementation. That is, on my machine --- a relatively fast 64-bit machine --- I can align two 400+ amino acid structures in about 0.300 s with the C++ implementation. In Python however, two 165 amino acid proteins took about 35 seconds!
When coupled to the Kabsch algorithm, this should be able to align any two protein structures, using just the alpha carbon coordinates.
This plugs into PyMol very easily. See the code and examples for installation and usage.
Comparison to PyMol
Why should you use this?
PyMol's structure alignment algorithm is fast and robust. However, its first step is to perform a sequence alignment of the two selections. Thus, proteins in the twilight zone or those having a low sequence identity, may not align well. Because CE is a structure-based alignment, this is not a problem. Look at the following example. The image at LEFT was the result of CE-aligning two proteins (1C0M to 1BCO). The result is 88 aligned (alpha carbons) residues (not atoms) at 2.78 Angstroms. The image on the RIGHT shows the results from PyMol's align command: an alignment of 221 atoms (not residues) at an RMSD of 15.7 Angstroms. To make the alignment easier to see, cealign (actually the Kabsch code) colors the aligned residues differently.
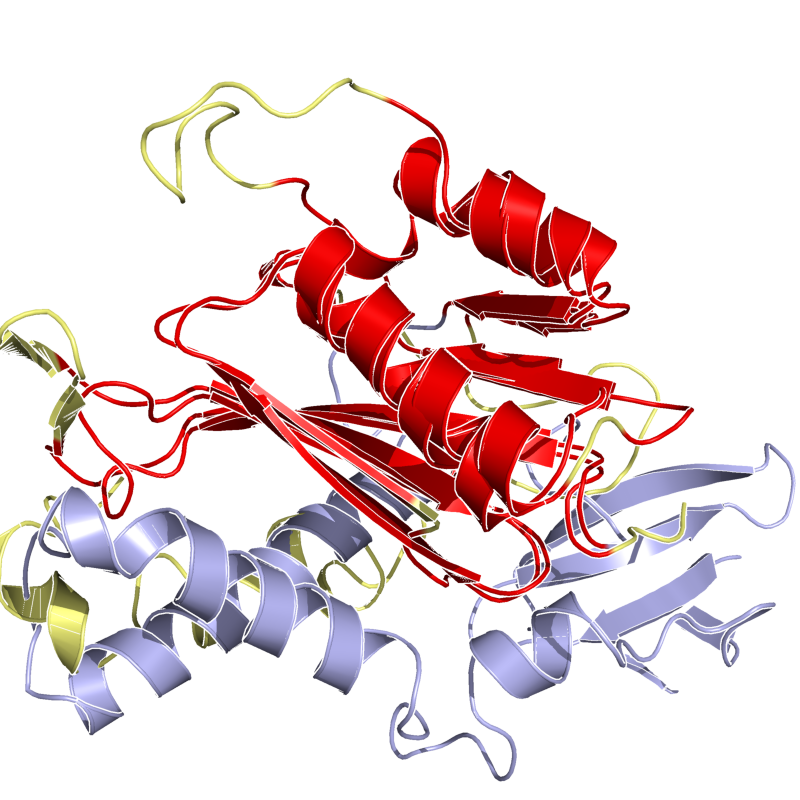
Cealign's results
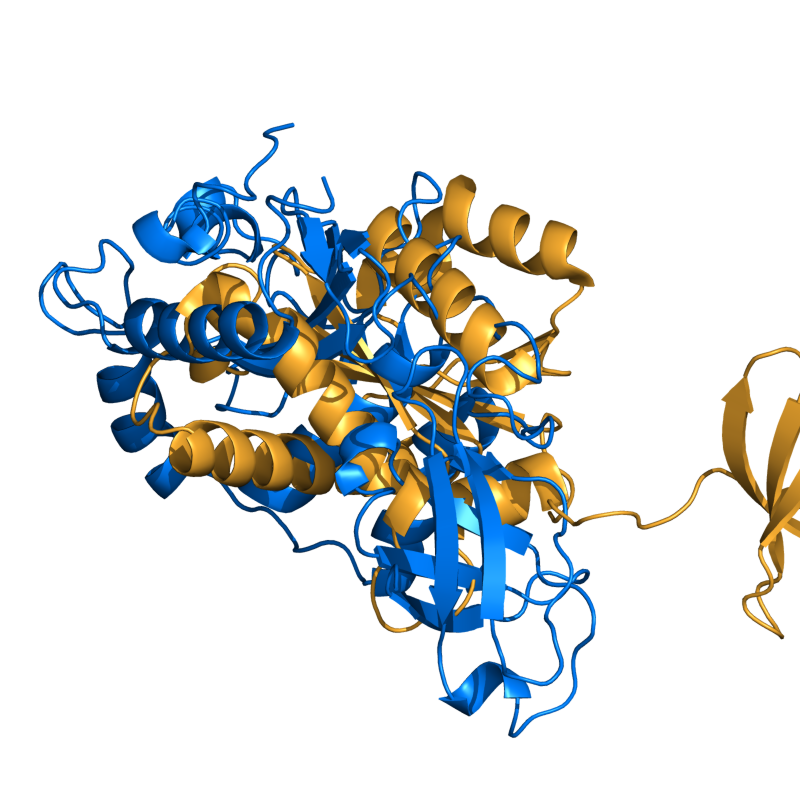
PyMol's results


Examples
Usage
Syntax
CEAlign has the semantic, and syntactic formalism of
cealign MASTER, TARGET
where a post-condition of the algorithm is that the coordinates of the MASTER protein are unchanged. This allows for easier multi-protein alignments. For example,
cealign 1AUE, 1BZ4
cealign 1AUE, 1B68
cealign 1AUE, 1A7V
cealign 1AUE, 1CPR
will superimpose all the TARGETS onto the MASTER.
Examples
cealign 1cll and i. 42-55, 1ggz and c. A
cealign 1kao, 1ctq
cealign 1fao, 1eaz
Multiple Structure Alignments
To use cealign to do a multiple structure alignment, simple load all your proteins and execute the following command:
for x in cmd.get_names("*"): cealign("MASTER", x)
where MASTER is the protein to align all others to. For example, load the following proteins: 1A15, 1EOT, 1ESR, 1F9R, 1G2S, 1NR4, 1QE6. Now, execute the command,
for x in cmd.get_names("*"): cealign("1A15", x)

Proteins Unaligned, just loaded into PyMOL.

All proteins aligned to 1A15


Paste the following code into the end of the cealign.py file that comes in the distribution. This function will define the alignto command in PyMOL. The function will align every object in PyMOL to the specified object.
def alignto(sel1):
"""Just a quick & dirty multiple structure alignment"""
for x in cmd.get_names("*"): cealign( sel1, x )
## Let PyMOL know about the alignto command
cmd.extend("alignto", alignto)
Once the code is in place, and PyMOL (or the cealign.py script) is reloaded, you can now execute,
alignto X
will align all objects in PyMOL to protein X. For example,
# for showing pretty representation
import preset
# get a bunch of similar structures and
# load them all at once.
fetch 1a2p 1bni 2f4y 2f56 2f5m 2f5w, async=0
# poor-man's multiple structure alignment
alignto 1bni
# make them 'pretty'.
preset.pretty("*")
# center the results (since they may all have
# moved out of frame).
center
Results From v0.2
Versions of CE Align, later than v0.2, should beat these alignments.
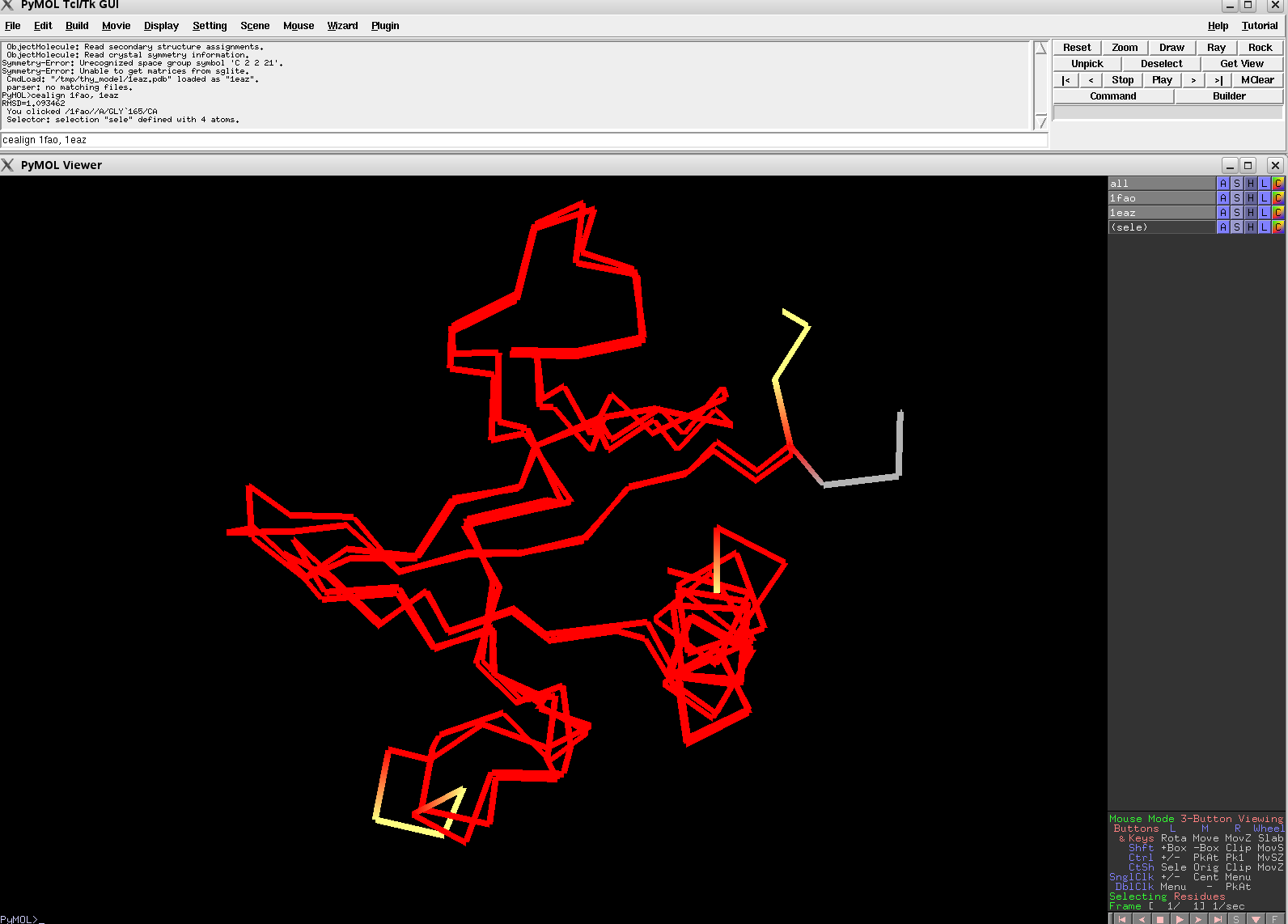
EASY: 1FAO vs. 1EAZ; 88 residues, 1.16 Ang
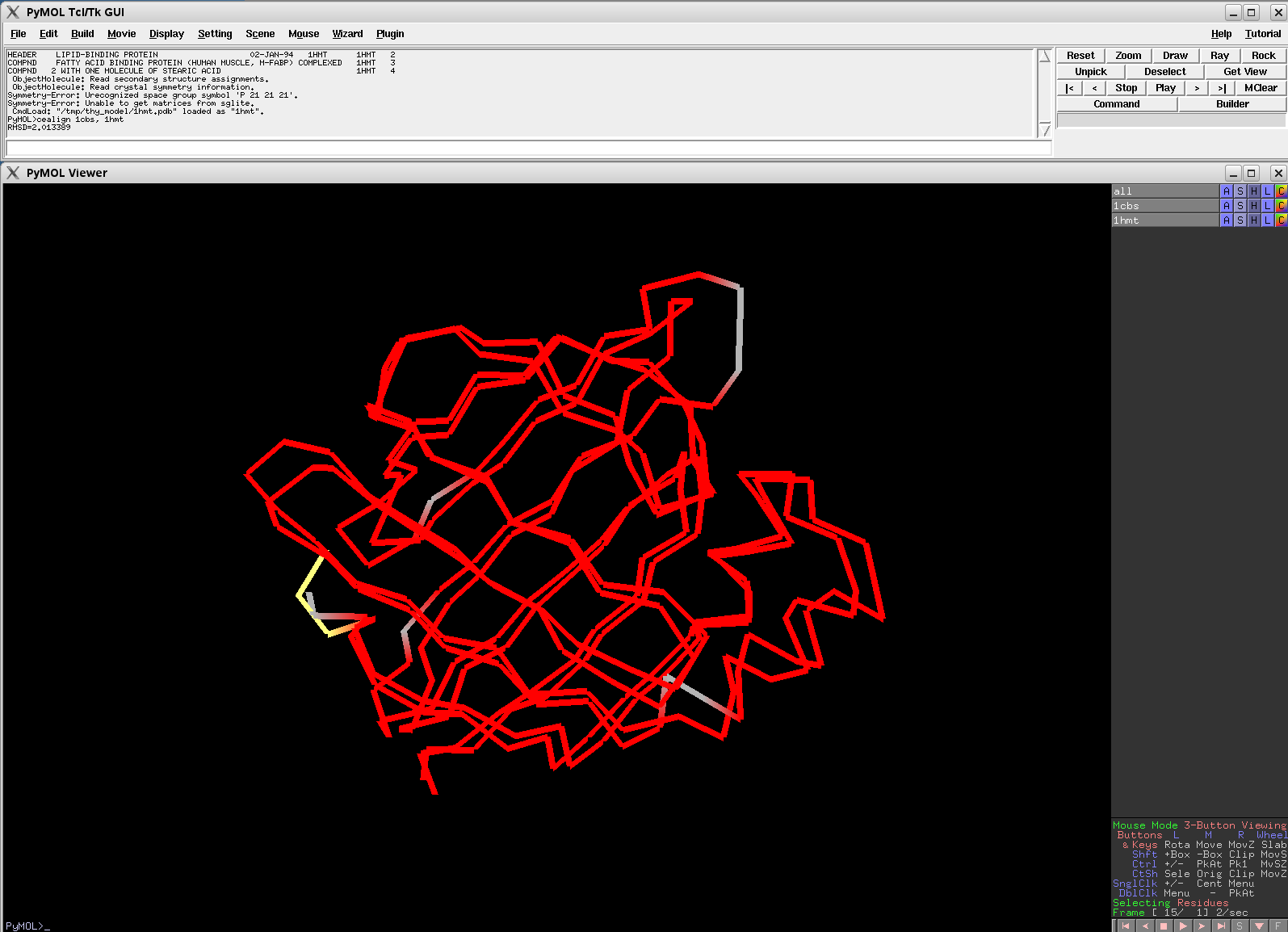
EASY: 1CBS vs. 1HMT; 120 residues, 2.07 Ang
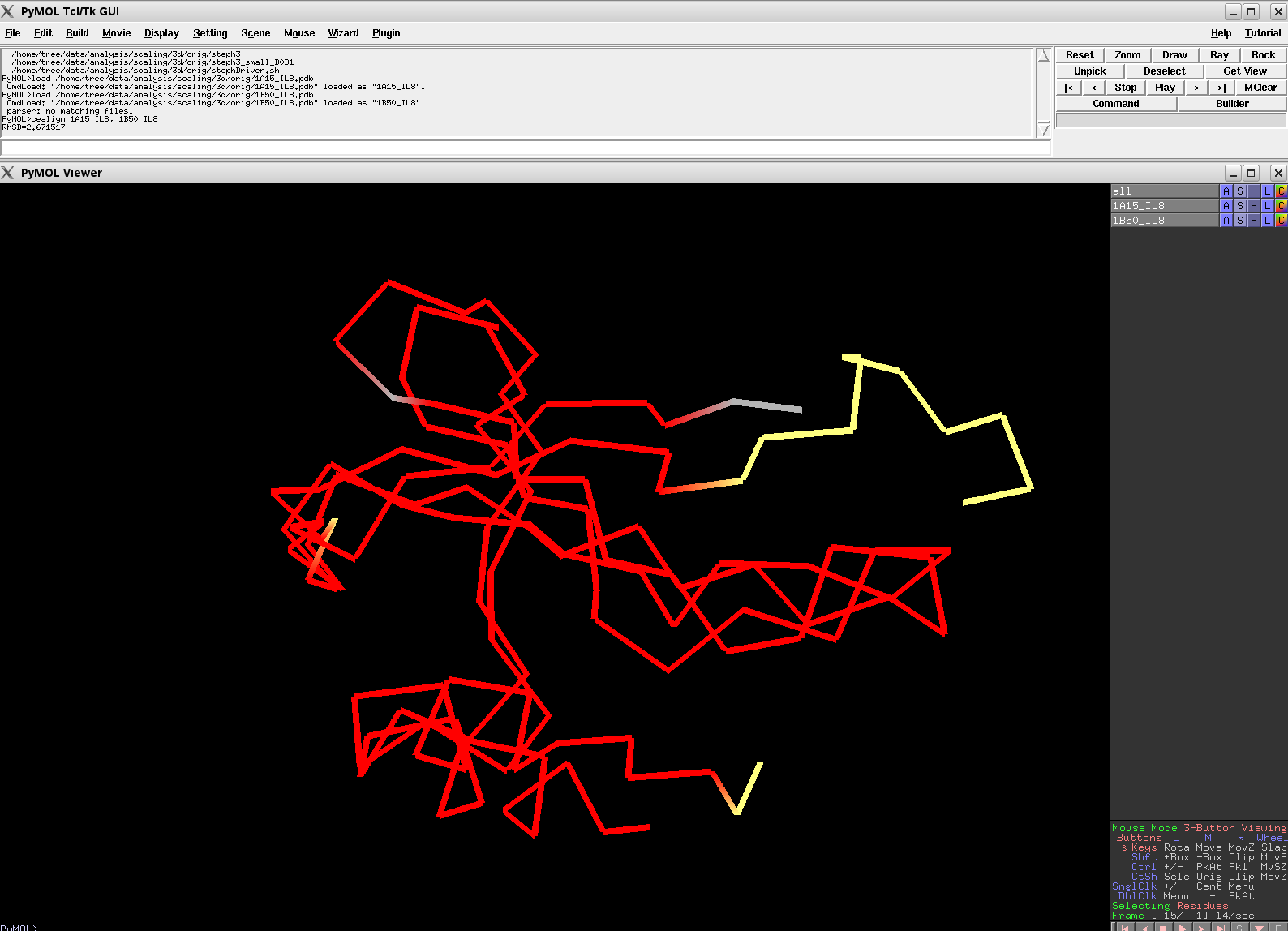
MODERATE: 1A15 vs 1B50; 56 residues, 6.67 Ang.
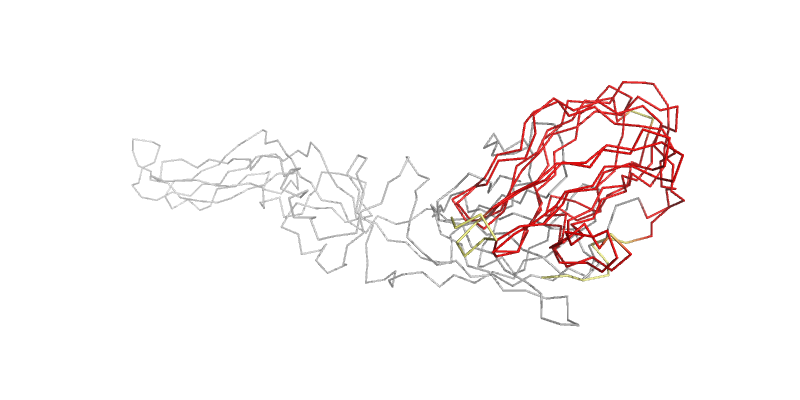
EASY: 1OAN vs. 1S6N; aligned to 2.26 Ang. RMSD.

HARD: 1RLW to 1BYN; 104 residues; 3.94 Ang.
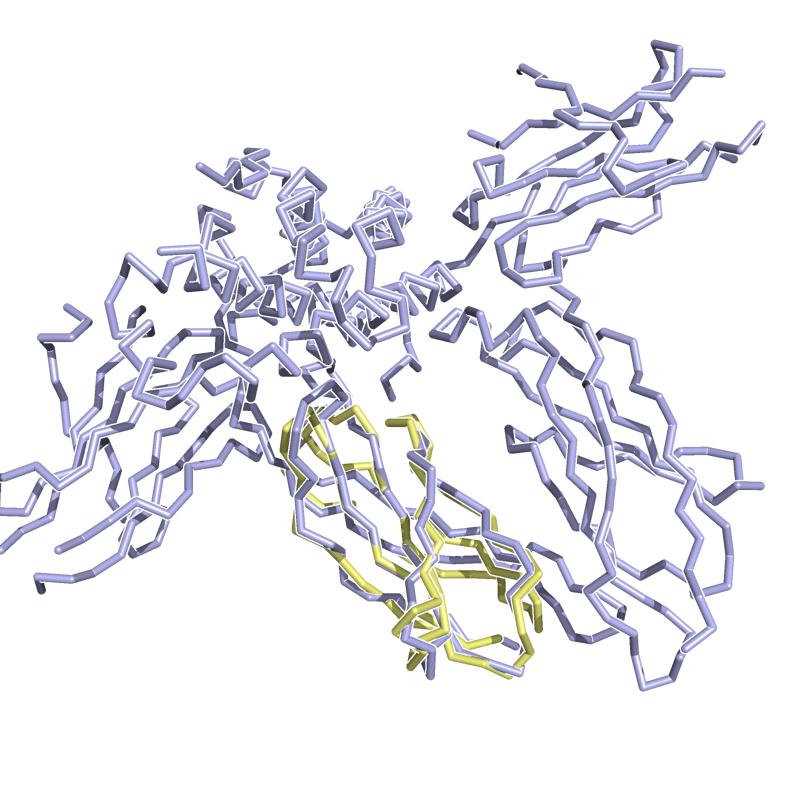
HARD: 1TEN vs. 3HHR; 72 residues, 3.13 Ang.
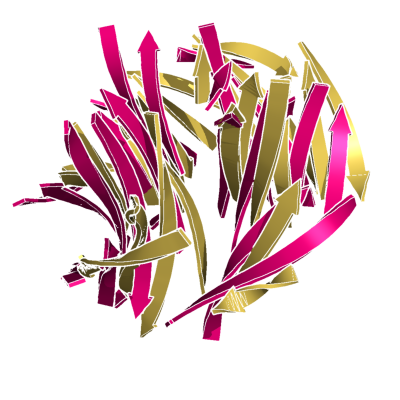
HARD: 2SIM vs. 1NSB; 280 residues, 5.00 Ang.
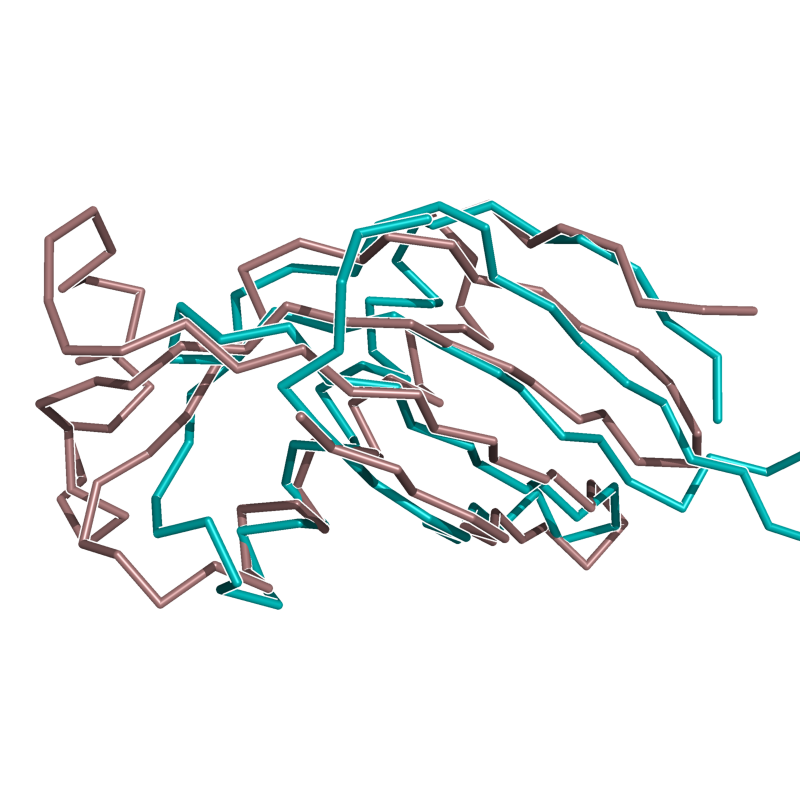
HARD: 1CEW vs. 1MOL; 72 residues, 3.63 Ang.








Installation
note: Windows installer coming soon.
Requirements
- Numpy
- Python 2.4+ with distutils
- C compiler
Directions
- uncompress the distribution file cealign-VERSION.tgz
- cd cealign-VERSION
- sudo python setup.py install
- insert "run DIR_TO_CEALIGN/cealign.py" and "run DIR_TO_CEALIGN/qkabsch.py" into your .pymolrc file, or just run the two Python scripts by hand.
- load some molecules
- run, cealign molecule1, molecule2
- enjoy
The Code
Please unpack and read the documentation. All comments/questions should be directed to Jason Vertrees (javertre _at_ utmb ...dot... edu).
LATEST IS v0.5.
Version 0.5
- BZ2 File CE Align v0.5
- ZIP File CE Align v0.5
Version 0.4
- BZ2 File CE Align v0.4
- ZIP File CE Align v0.4
Version 0.3
- BZ2 File CE Align v0.3
- ZIP File CE Align v0.3
Version 0.2
- BZ2 File CE Align v0.2
- ZIP File CE Align v0.2
Version 0.1
- BZ2 File CE Align v0.1
- ZIP File CE Align v0.1
Updates
2007-02-20
A HUGE thanks to Dan Kulp' for spotting the bug in the reflections! I have fixed the code and repackaged it. Briefly, I found bug in the way Numpy was returning values from its' det function. It was returning something that was not casting correctly to Python's internal types. For example, the determinant of an orthonormal matrix must be +/- 1.0 -- and hence the product of two orthonormal matrices must also be +/- 1.0 -- but here were the test results from Numpy:
Reflect => -1.0
Does reflect equal 1.0 => False
Reflect => -1.0
Does reflect equal -1.0 =>False
The second case is obviously wrong, and the program was never properly detecting reflections. The fix is:
reflect = float(str(float(numpy.linalg.det(V) * numpy.linalg.det(Wt))))
if reflect == -1.0:
S[-1] = -S[-1]
V[:,-1] = -V[:,-1]
2007-02-01
Argh! Found another stupid bug. The alignments are now longer and more accurate. v0.4 soon.
2007-01-31
Found a small bug. Will update code soon.
2007-01-25
CE Align v0.2 released.
Found a "feature" I don't like; so, I fixed it. The new version of cealign has the formal syntax of
cealign MASTER, TARGET
and cealign is now guaranteed not to change the coordinates of the MASTER protein. This is useful is you want to align 10 structures on top of one. Before, cealign would center the two molecules; now it just overlaps the TARGET onto the MASTER.
2007-01-17
CE Align V0.1 released.
2007-01-11
The first version of the C-module code is complete. I fixed handling (multiple) missing residues, the centering problem, and the problem of multiple chains. I'll package and provide the code soon.
2007-01-10
Trying to remedy missing residues. If a user's selections are protA and i. 10-20 and prot2 and i. 10-20, and if prot2 is missing residue 14, the SVD is undefined/inappropriate. I have to weed out residues that don't have partners in the PDB file. Alignments do this implicitly since the XYZ values it sees are only the ones with coordinates. Also, CE only works on individual chains. If someone can find a consistent method to map residues and chains to ints and then back to residues and chains -- that might work. Ha!
If more than a week lapses after this comment, I'll just wrap up the code and post the first version. There seems to be some interest in this plugin, so the more eyes the easier it may be to fix the bugs. I will also need testers for the Mac and Windows editions.
2007-01-08
Yeah! The C code that plugs into PyMol has been completed. It's a little slower than the plain C++ code I wrote, but that's what you get when passing data from PyMol to Python to C, fiddle with it, pass it back to Python to PyMol for some more quick math. The alignment times for the two proteins mentioned below (1B50 and 1C0M) on my machine with the new C module is about 1-3 second (with a full CPU load for other intensive tasks running in the background; this shows great improvement over the pure Python alignment times). Once the code is cleaned up (and I'm not too embarrassed to post it) and some bugs are worked out, I'll post it. The current bugs are:
- Some alignments don't center right
- Missing residues cause problems
- Memory leaks galore, I'm sure
The code consists of:
- qkabsch.py
- cealign.py
- ccealignmodule.c
- ccealignmodule.h
- setup.py
Also, I provide the option of aligning based solely upon RMSD or upon the better CE-Score. See the References for information on the CE Score.
Troubleshooting
Post your problems/solutions here.
LinAlg Module Not Found
Problem: Running CE Align gives the following error message:
run qkabsch.py
Traceback (most recent call last):
File "/usr/lib/python2.4/site-packages/pymol/parser.py", line 285, in parse
parsing.run_file(exp_path(args[nest][0]),pymol_names,pymol_names)
File "/usr/lib/python2.4/site-packages/pymol/parsing.py", line 407, in run_file
execfile(file,global_ns,local_ns)
File "qkabsch.py", line 86, in ?
import numpy
File "/usr/lib/python2.4/site-packages/numpy/__init__.py", line 40, in ?
import linalg
ImportError: No module named linalg
Solution: You do not have the linear algebra module installed (or Python can't find it) on your machine. One workaround is to install Scientific Python. Another is to reinstall the Numpy package from source, ensuring that you have the necessary requirements for the linear algebra module (linpack, lapack, fft, etc.).
CCEAlign & NumPy Modules Not Found
Problem: Running CE Align gives the following error message:
PyMOL>run cealign.py
Traceback (most recent call last):
File "/home/local/warren/MacPyMOL060530/build/Deployment/MacPyMOL.app/pymol/modules/pymol/parser.py", line 297, in parse
File "/home/local/warren/MacPyMOL060530/build/Deployment/MacPyMOL.app/pymol/modules/pymol/parsing.py", line 408, in run_file
File "/usr/local/pymol/scripts/cealign-0.1/cealign.py", line 59, in ?
from ccealign import ccealign
ImportError: No module named ccealign
run qkabsch.py
Traceback (most recent call last):
File "/home/local/warren/MacPyMOL060530/build/Deployment/MacPyMOL.app/pymol/modules/pymol/parser.py", line 297, in parse
File "/home/local/warren/MacPyMOL060530/build/Deployment/MacPyMOL.app/pymol/modules/pymol/parsing.py", line 408, in run_file
File "qkabsch.py", line 86, in ?
import numpy
ImportError: No module named numpy
Solution: This problem occurs under Apple Mac OS X if (a) the Apple's python executable on your machine (/usr/bin/python, currently version 2.3.5) is superseded by Fink's python executable (/sw/bin/python, currently version 2.5) and (b) you are using precompiled versions of PyMOL (MacPyMOL, PyMOLX11Hybrid or PyMOL for Mac OS X/X11). These executables ignore Fink's python and instead use Apple's - so, in order to run CE Align, one must install NumPy (as well as CE Align itself) using Apple's python. To do so, first download the Numpy source code archive (currently version 1.0.1), unpack it, change directory to numpy-1.0.1 and specify the full path to Apple's python executable during installation: sudo /usr/bin/python setup.py install | tee install.log. Then, donwload the CE Align source code archive (currently version 0.2), unpack it, change directory to cealign-0.2 and finally install CE Align as follows: sudo /usr/bin/python setup.py install | tee install.log. Luca Jovine 05:11, 25 January 2007 (CST).
References
Text taken from PubMed and formatted for the wiki. The first reference is the most important for this code.
- Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998 Sep;11(9):739-47. PMID: 9796821 [PubMed - indexed for MEDLINE]
- Jia Y, Dewey TG, Shindyalov IN, Bourne PE. A new scoring function and associated statistical significance for structure alignment by CE. J Comput Biol. 2004;11(5):787-99. PMID: 15700402 [PubMed - indexed for MEDLINE]
- Pekurovsky D, Shindyalov IN, Bourne PE. A case study of high-throughput biological data processing on parallel platforms. Bioinformatics. 2004 Aug 12;20(12):1940-7. Epub 2004 Mar 25. PMID: 15044237 [PubMed - indexed for MEDLINE]
- Shindyalov IN, Bourne PE. An alternative view of protein fold space. Proteins. 2000 Feb 15;38(3):247-60. PMID: 10713986 [PubMed - indexed for MEDLINE]
License
The CEAlign and all its subprograms that I wrote, are released under the open source Free BSD License (BSDL).