|
|
| (17 intermediate revisions by one other user not shown) |
| Line 1: |
Line 1: |
| == Acknowledgement == | | {{Infobox script-repo |
| This pymol script is made by Troels Emtekær Linnet.
| | |type = script |
| | | |filename = scripts/colorbydisplacement.py |
| This script is based on the scaffold from [[ColorByRMSD]]. Peace love and harmony goes to Shivender Shandilya and Jason Vertrees.
| | |author = [[User:Tlinnet|Troels E. Linnet]] |
| | |license = BSD |
| | }} |
|
| |
|
| == Introduction == | | == Introduction == |
| This script allows you to color two structures by distance displacement between an Open and Closed form of a protein, as calculated by PyMol's internal distance command. The pairwise distance is calculated between, C-alpha or all-atoms. The distance displacement values are stored as B-factors of these residues, which are colored by a ''rainbow'' color spectrum, with blue specifying minimum and red indicating maximum. | | This script allows you to color two structures by distance displacement between an Open and Closed form of a protein, as calculated by PyMol's internal distance command. The pairwise distance is calculated between all-atoms. The distance displacement values are stored as B-factors of these residues, which are colored by a ''rainbow'' color spectrum, with blue specifying minimum and red indicating maximum. |
|
| |
|
| == Code ==
| |
| Do keep in mind, all original B-factors values are overwritten! | | Do keep in mind, all original B-factors values are overwritten! |
|
| |
|
| There exist two versions. <br> | | There exist one version. <br> |
| ColorByDisplacement'''CA''' is quick and is between CA atoms. Ideal for helices representation. <br>
| |
| ColorByDisplacement'''All''' is between All atoms in residues and is quite slow => 3-5 mins for a run. Ideal for sticks representation. | | ColorByDisplacement'''All''' is between All atoms in residues and is quite slow => 3-5 mins for a run. Ideal for sticks representation. |
|
| |
|
| Line 24: |
Line 24: |
|
| |
|
| === Examples === | | === Examples === |
| <source lang="python"> | | <syntaxhighlight lang="python"> |
| ColorByDisplacementCA O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t
| |
| ColorByDisplacementAll O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t | | ColorByDisplacementAll O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t |
|
| |
|
| ColorByDisplacementCA O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t, AlignedWhite='no'
| |
| ColorByDisplacementAll O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t, AlignedWhite='no' | | ColorByDisplacementAll O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t, AlignedWhite='no' |
| | | </syntaxhighlight> |
| ColorByDisplacementCA O5NT, C5NT, resi 26-355, resi 26-355
| |
| ColorByDisplacementAll O5NT, C5NT, resi 26-355, resi 26-355
| |
| </source> | |
|
| |
|
| <gallery heights="240px" widths="340px"> | | <gallery heights="240px" widths="340px"> |
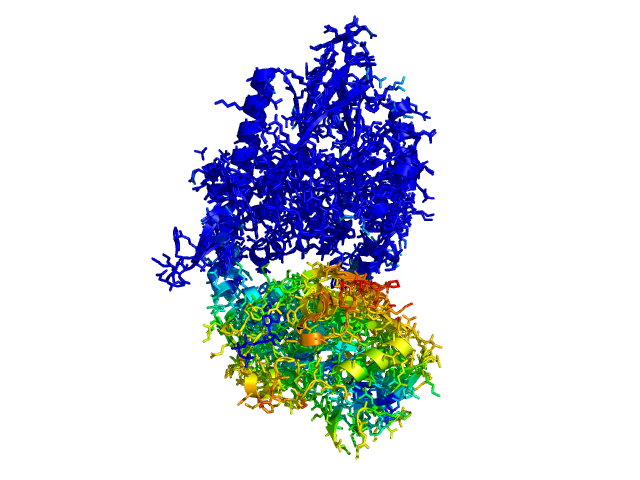
| Image:ColorByDisplacement-CA-1.png|ColorByDisplacementCA used on 1HP1 and 1HPU aligned and colored by distance displacement.
| |
| Image:ColorByDisplacement-CA-2.png|ColorByDisplacementCA used on 1HP1 and 1HPU aligned and colored by distance displacement.
| |
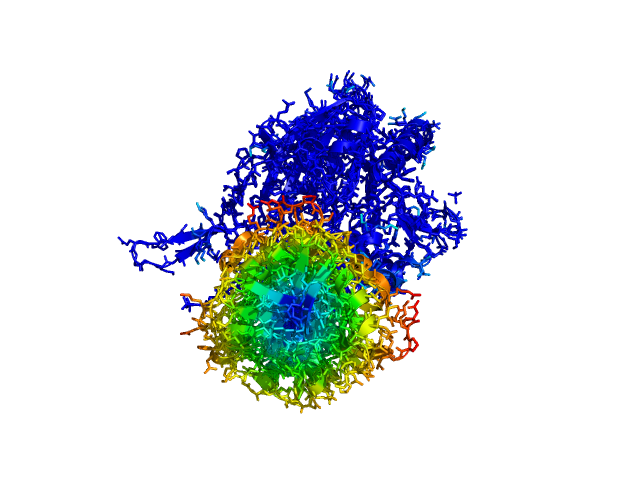
| Image:ColorByDisplacement-All-1.png|ColorByDisplacementAll used on 1HP1 and 1HPU aligned and colored by distance displacement. | | Image:ColorByDisplacement-All-1.png|ColorByDisplacementAll used on 1HP1 and 1HPU aligned and colored by distance displacement. |
| Image:ColorByDisplacement-All-2.png|ColorByDisplacementAll used on 1HP1 and 1HPU aligned and colored by distance displacement. | | Image:ColorByDisplacement-All-2.png|ColorByDisplacementAll used on 1HP1 and 1HPU aligned and colored by distance displacement. |
| Line 45: |
Line 38: |
| Residues not in both pdb files is colored black | | Residues not in both pdb files is colored black |
|
| |
|
| == Example Pymol Script == | | == Example 1 == |
| <source lang="python">
| | {{Template:PymolScriptRepoDownload|examples/colorbydisplacement_1.pml}} |
| cd C:\Users\tlinnet\Documents\My Dropbox\Speciale\5NT-project\Mutant-construct\Distance-Plot
| | <include src="https://raw.github.com/Pymol-Scripts/Pymol-script-repo/master/examples/colorbydisplacement_1.pml" highlight="python" /> |
| #cd /homes/linnet/Documents/Speciale/5NT-project/Mutant-construct/Distance-Plot
| |
| ### load pdb files and rename
| |
| load 1HP1.pdb, O5NT-1HP1
| |
| load 1HPU.pdb, C5NT-1HPU
| |
| | |
| hide everything
| |
| ### Select asymmetric units from pdb file
| |
| create O5NT, /O5NT-1HP1//A
| |
| create C5NT, /C5NT-1HPU//C
| |
| delete O5NT-1HP1
| |
| delete C5NT-1HPU
| |
| | |
| cartoon auto
| |
| show cartoon, O5NT
| |
| show cartoon, C5NT
| |
| set cartoon_fancy_helices=1
| |
| set bg,[1,1,1]
| |
| | |
| set auto_zoom, off
| |
| | |
| ### Make sharper
| |
| set fog=0
| |
| | |
| ### Load my function, and run function with input
| |
| run ColorByDisplacement.py
| |
| ColorByDisplacementCA O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t
| |
| #ColorByDisplacementAll O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t
| |
| | |
| set_view (\
| |
| 0.094686687, -0.390707940, 0.915631354,\
| |
| 0.809000611, -0.505792081, -0.299485058,\
| |
| 0.580131471, 0.769104064, 0.268191338,\
| |
| 0.000000000, 0.000000000, -280.940521240,\
| |
| 26.240486145, 46.146961212, 21.702068329,\
| |
| 231.830673218, 330.050415039, -20.000000000 )
| |
| </source>
| |
| | |
| | |
| == Python Code ==
| |
| <source lang="python"> | |
| import pymol
| |
| import cmd
| |
| from pymol import stored
| |
| | |
| ### Thanks for inspiration from:
| |
| """
| |
| --- ColorByRMSD: RMSD based coloring ---
| |
| Authors : Shivender Shandilya; Jason Vertrees
| |
| Program : ColorByRMSD
| |
| Date : July 2009
| |
| http://www.pymolwiki.org/index.php/ColorByRMSD
| |
| """
| |
| ### Author Troels Linnet - troels.linnet att bbz.uni-leipzig.de
| |
| """
| |
| --- ColorByDisplacementCA: Displacement based coloring ---
| |
| Authors : Troels E. Linnet
| |
| Program : ColorByDisplacementCA
| |
| Date : January 2011
| |
| email: troels.linnet att bbz.uni-leipzig.de
| |
| """
| |
| | |
| """
| |
| ColorByDisplacementCA --
| |
| Show the distance displacement deviation in color to more easily see variable regions.
| |
| | |
| PARAMS
| |
| | |
| objSel1 (valid PyMOL object or selection)
| |
| The first object
| |
| | |
| objSel2 (valid PyMOL object or selection)
| |
| The second object
| |
| | |
| doColor (boolean, either True or False)
| |
| If doColor=True then a simple representation is created to
| |
| highlight the differences. If False, then no changes are made.
| |
| DEFAULT: False
| |
| | |
| RETURNS
| |
| None.
| |
| | |
| SIDE-EFFECTS
| |
| Modifies the B-factor columns in your original structures.
| |
| | |
| """
| |
| | |
| def strTrue(p):
| |
| return p[0].upper() == "T"
| |
| | |
| # The main function that assigns current displacement distance as the new B-factor
| |
| def displacementUpdateB(objA, alnAri, objB, alnBri):
| |
| ### If residue is unassigned in one of the pdb files, we reset its value
| |
| for x in range(len(alnAri)):
| |
| s1 = objA + " and name CA and resi " + alnAri[x]
| |
| cmd.alter( s1, "b = " + str(-0.01))
| |
| for x in range(len(alnBri)):
| |
| s2 = objB + " and name CA and resi " + alnBri[x]
| |
| cmd.alter( s2, "b = " + str(-0.01))
| |
| cmd.sort(objA); cmd.sort(objB)
| |
| for x in range(len(alnAri)):
| |
| s1 = objA + " and name CA and resi " + alnAri[x]
| |
| s2 = objB + " and name CA and resi " + alnAri[x]
| |
| ### Names starting with __ (underscores) are normally hidden by PyMOL
| |
| tempObject = "__tempObject"
| |
| Displacement = cmd.distance(tempObject, s1, s2)
| |
| cmd.alter( s1, "b = " + str(Displacement))
| |
| cmd.alter( s2, "b = " + str(Displacement))
| |
| cmd.delete(tempObject)
| |
| cmd.sort(objA); cmd.sort(objB)
| |
| | |
| def ColorByDisplacementCA(objSel1, objSel2, super1='all', super2='all', doColor="True", doAlign="True", AlignedWhite='yes'):
| |
| ### First create backup copies; names starting with __ (underscores) are normally hidden by PyMOL
| |
| tObj1, tObj2, aln = "__tempObj1", "__tempObj2", "__aln"
| |
| | |
| if strTrue(doAlign):
| |
| ### Create temp objects
| |
| cmd.create( tObj1, objSel1 )
| |
| cmd.create( tObj2, objSel2 )
| |
| ### Align and make create an object aln which indicates which atoms were paired between the two structures
| |
| ### Super is must faster than align http://www.pymolwiki.org/index.php/Super
| |
| cmd.super(tObj1 + ' and ' + str(super1), tObj2 + ' and ' + str(super2), object=aln)
| |
| ### Modify the original matrix of object1 from the alignment
| |
| cmd.matrix_copy(tObj1, objSel1)
| |
| else:
| |
| ### Create temp objects
| |
| cmd.create( tObj1, objSel1 )
| |
| cmd.create( tObj2, objSel2 )
| |
| ### Align and make create an object aln which indicates which atoms were paired between the two structures
| |
| ### Super is must faster than align http://www.pymolwiki.org/index.php/Super
| |
| cmd.super(tObj1 + ' and ' + str(super1), tObj2 + ' and ' + str(super2), object=aln)
| |
| | |
| ### Modify the B-factor columns of the original objects,
| |
| ### in order to identify the residues NOT used for alignment, later on
| |
| cmd.alter( objSel1 + " or " + objSel2, "b=-0.2")
| |
| cmd.alter( tObj1 + " or " + tObj2, "chain='A'")
| |
| cmd.alter( tObj1 + " or " + tObj2, "segi='A'")
| |
| | |
| ### Update pymol internal representations; one of these should do the trick
| |
| cmd.refresh(); cmd.rebuild(); cmd.sort(tObj1); cmd.sort(tObj2)
| |
| | |
| ### Create lists for storage
| |
| stored.alnAres, stored.alnBres = [], []
| |
| | |
| ### Iterate over objects
| |
| if AlignedWhite=='yes':
| |
| cmd.iterate(tObj1 + " and n. CA and not " + aln, "stored.alnAres.append(resi)")
| |
| cmd.iterate(tObj2 + " and n. CA and not " + aln, "stored.alnBres.append(resi)")
| |
| else:
| |
| cmd.iterate(tObj1 + " and n. CA", "stored.alnAres.append(resi)")
| |
| cmd.iterate(tObj2 + " and n. CA", "stored.alnBres.append(resi)")
| |
| | |
| ### Change the B-factors for EACH object
| |
| displacementUpdateB(tObj1,stored.alnAres,tObj2,stored.alnBres)
| |
| | |
| ### Store the NEW B-factors
| |
| stored.alnAnb, stored.alnBnb = [], []
| |
| ### Iterate over objects and get b
| |
| if AlignedWhite=='yes':
| |
| ### Iterate over objects which is not aligned
| |
| cmd.iterate(tObj1 + " and n. CA and not " + aln, "stored.alnAnb.append(b)" )
| |
| cmd.iterate(tObj2 + " and n. CA and not " + aln, "stored.alnBnb.append(b)" )
| |
| else:
| |
| ### Or Iterate over all objects with CA
| |
| cmd.iterate(tObj1 + " and n. CA", "stored.alnAnb.append(b)" )
| |
| cmd.iterate(tObj2 + " and n. CA", "stored.alnBnb.append(b)" )
| |
| | |
| ### Get rid of all intermediate objects and clean up
| |
| cmd.delete(tObj1)
| |
| cmd.delete(tObj2)
| |
| cmd.delete(aln)
| |
| | |
| ### Assign the just stored NEW B-factors to the original objects
| |
| for x in range(len(stored.alnAres)):
| |
| cmd.alter(objSel1 + " and n. CA and i. " + str(stored.alnAres[x]), "b = " + str(stored.alnAnb[x]))
| |
| for x in range(len(stored.alnBres)):
| |
| cmd.alter(objSel2 + " and n. CA and i. " + str(stored.alnBres[x]), "b = " + str(stored.alnBnb[x]))
| |
| cmd.rebuild(); cmd.refresh(); cmd.sort(objSel1); cmd.sort(objSel2)
| |
| | |
| ### Provide some useful information
| |
| stored.allRMSDval = []
| |
| stored.allRMSDval = stored.alnAnb + stored.alnBnb
| |
| print "\nColorByDisplacementCA completed successfully."
| |
| print "The MAXIMUM Displacement is: "+str(max(stored.allRMSDval)) +" residue "+str(stored.alnAres[int(stored.allRMSDval.index(max(stored.allRMSDval)))])
| |
| | |
| if strTrue(doColor):
| |
| ### Showcase what we did
| |
| #cmd.orient()
| |
| #cmd.hide("all")
| |
| cmd.show("cartoon", objSel1 + " or " + objSel2)
| |
| ### Select the residues not used for alignment; they still have their B-factors as "-0.2"
| |
| cmd.select("notUsedForAln", "b = -0.2")
| |
| ### White-wash the residues not used for alignment
| |
| cmd.color("white", "notUsedForAln")
| |
| ### Select the residues not in both pdb files; they have their B-factors as "-0. 01"
| |
| cmd.select("ResNotInBothPDB", "b = -0.01")
| |
| ### White-wash the residues not used for alignment
| |
| cmd.color("black", "ResNotInBothPDB")
| |
| ### Color the residues used for alignment according to their B-factors (Displacment values)
| |
| # cmd.spectrum("b", 'rainbow', "((" + objSel1 + " and n. CA) or (n. CA and " + objSel2 +" )) and not notUsedForAln+ResNotInBothPDB")
| |
| cmd.spectrum("b", 'rainbow', "((" + objSel1 + " and n. CA) or (n. CA and " + objSel2 +" )) and not (notUsedForAln or ResNotInBothPDB)")
| |
| ### Delete the selection of atoms not used for alignment
| |
| ### If you would like to keep this selection intact,
| |
| ### just comment "cmd.delete" line and
| |
| ### uncomment the "cmd.disable" line abowe.
| |
| cmd.disable("notUsedForAln")
| |
| cmd.delete("notUsedForAln")
| |
| cmd.disable("ResNotInBothPDB")
| |
| cmd.delete("ResNotInBothPDB")
| |
| | |
| print "\nObjects are now colored by C-alpha displacement deviation."
| |
| print "Blue is minimum and red is maximum..."
| |
| print "White is those residues used in the alignment algorithm. Can be turned off in top of algorithm."
| |
| print "Black is residues that does not exist in both files..."
| |
| cmd.extend("ColorByDisplacementCA", ColorByDisplacementCA)
| |
| | |
| def displacementUpdateBAll(objA, alnAri, objB, alnBri):
| |
| print "This will take a while to go through the for loops. Give me around 3-5 minutes..."
| |
| ### If residue is unassigned in one of the pdb files, we reset its value
| |
| for x in range(len(alnAri)):
| |
| s1 = objA + " and resi " + alnAri[x][0] + " and name " + str(alnAri[x][1])
| |
| cmd.alter( s1, "b = " + str(-0.01))
| |
| for x in range(len(alnBri)):
| |
| s2 = objB + " and resi " + alnBri[x][0] + " and name " + alnBri[x][1]
| |
| cmd.alter( s2, "b = " + str(-0.01))
| |
| cmd.sort(objA); cmd.sort(objB)
| |
| for x in range(len(alnAri)):
| |
| s1 = objA + " and resi " + alnAri[x][0] + " and name " + alnAri[x][1]
| |
| s2 = objB + " and resi " + alnAri[x][0] + " and name " + alnAri[x][1]
| |
| ### Names starting with __ (underscores) are normally hidden by PyMOL
| |
| tempObject = "__tempObject"
| |
| Displacement = cmd.distance(tempObject, s1, s2)
| |
| cmd.alter( s1, "b = " + str(Displacement))
| |
| cmd.alter( s2, "b = " + str(Displacement))
| |
| cmd.delete(tempObject)
| |
| cmd.sort(objA); cmd.sort(objB)
| |
| | |
| def ColorByDisplacementAll(objSel1, objSel2, super1='all', super2='all', doColor="True", doAlign="True", AlignedWhite='yes'):
| |
| ### First create backup copies; names starting with __ (underscores) are normally hidden by PyMOL
| |
| tObj1, tObj2, aln = "__tempObj1", "__tempObj2", "__aln"
| |
| | |
| if strTrue(doAlign):
| |
| ### Create temp objects
| |
| cmd.create( tObj1, objSel1 )
| |
| cmd.create( tObj2, objSel2 )
| |
| ### Align and make create an object aln which indicates which atoms were paired between the two structures
| |
| ### Super is must faster than align http://www.pymolwiki.org/index.php/Super
| |
| cmd.super(tObj1 + ' and ' + str(super1), tObj2 + ' and ' + str(super2), object=aln)
| |
| ### Modify the original matrix of object1 from the alignment
| |
| cmd.matrix_copy(tObj1, objSel1)
| |
| else:
| |
| ### Create temp objects
| |
| cmd.create( tObj1, objSel1 )
| |
| cmd.create( tObj2, objSel2 )
| |
| ### Align and make create an object aln which indicates which atoms were paired between the two structures
| |
| ### Super is must faster than align http://www.pymolwiki.org/index.php/Super
| |
| cmd.super(tObj1 + ' and ' + str(super1), tObj2 + ' and ' + str(super2), object=aln)
| |
| | |
| ### Modify the B-factor columns of the original objects,
| |
| ### in order to identify the residues NOT used for alignment, later on
| |
| cmd.alter( objSel1 + " or " + objSel2, "b=-0.2")
| |
| cmd.alter( tObj1 + " or " + tObj2, "chain='A'")
| |
| cmd.alter( tObj1 + " or " + tObj2, "segi='A'")
| |
| | |
| ### Update pymol internal representations; one of these should do the trick
| |
| cmd.refresh(); cmd.rebuild(); cmd.sort(tObj1); cmd.sort(tObj2)
| |
| | |
| ### Create lists for storage
| |
| stored.alnAres, stored.alnBres = [], []
| |
| | |
| ### Iterate over objects and get resi
| |
| if AlignedWhite=='yes':
| |
| cmd.iterate(tObj1 + " and not " + aln, "stored.alnAres.append((resi, name))")
| |
| cmd.iterate(tObj2 + " and not " + aln, "stored.alnBres.append((resi, name))")
| |
| else:
| |
| cmd.iterate(tObj1, "stored.alnAres.append((resi, name))")
| |
| cmd.iterate(tObj2, "stored.alnBres.append((resi, name))")
| |
| | |
| ### Change the B-factors for EACH object
| |
| displacementUpdateBAll(tObj1,stored.alnAres,tObj2,stored.alnBres)
| |
| | |
| ### Store the NEW B-factors
| |
| stored.alnAnb, stored.alnBnb = [], []
| |
| ### Iterate over objects and get b
| |
| | |
| if AlignedWhite=='yes':
| |
| ### Iterate over objects which is not aligned
| |
| cmd.iterate(tObj1 + " and not " + aln, "stored.alnAnb.append(b)" )
| |
| cmd.iterate(tObj2 + " and not " + aln, "stored.alnBnb.append(b)" )
| |
| else:
| |
| ### Or Iterate over all objects with CA
| |
| cmd.iterate(tObj1, "stored.alnAnb.append(b)" )
| |
| cmd.iterate(tObj2, "stored.alnBnb.append(b)" )
| |
| | |
| ### Get rid of all intermediate objects and clean up
| |
| cmd.delete(tObj1)
| |
| cmd.delete(tObj2)
| |
| cmd.delete(aln)
| |
| | |
| ### Assign the just stored NEW B-factors to the original objects
| |
| print "Sooon ready. 1 more minute"
| |
| for x in range(len(stored.alnAres)):
| |
| cmd.alter(objSel1 + " and resi " + str(stored.alnAres[x][0]) + " and name " + str(stored.alnAres[x][1]), "b = " + str(stored.alnAnb[x]))
| |
| for x in range(len(stored.alnBres)):
| |
| cmd.alter(objSel2 + " and resi " + str(stored.alnBres[x][0]) + " and name " + str(stored.alnBres[x][1]), "b = " + str(stored.alnBnb[x]))
| |
| cmd.rebuild(); cmd.refresh(); cmd.sort(objSel1); cmd.sort(objSel2)
| |
| | |
| ### Provide some useful information
| |
| stored.allRMSDval = []
| |
| stored.allRMSDval = stored.alnAnb + stored.alnBnb
| |
| print "\nColorByDisplacementAll completed successfully."
| |
| print "The MAXIMUM Displacement is: "+str(max(stored.allRMSDval)) +" residue "+str(stored.alnAres[int(stored.allRMSDval.index(max(stored.allRMSDval)))])
| |
| | |
| if strTrue(doColor):
| |
| ### Showcase what we did
| |
| #cmd.orient()
| |
| #cmd.hide("all")
| |
| cmd.show("sticks", objSel1 + " or " + objSel2)
| |
| ### Select the residues not used for alignment; they still have their B-factors as "-0.2"
| |
| cmd.select("notUsedForAln", "b = -0.2")
| |
| ### White-wash the residues not used for alignment
| |
| cmd.color("white", "notUsedForAln")
| |
| ### Select the residues not in both pdb files; they have their B-factors as "-0.01"
| |
| cmd.select("ResNotInBothPDB", "b = -0.01")
| |
| ### White-wash the residues not used for alignment
| |
| cmd.color("black", "ResNotInBothPDB")
| |
| ### Color the residues used for alignment according to their B-factors (Displacement values)
| |
| # cmd.spectrum("b", 'rainbow', "((" + objSel1 + ") or (" + objSel2 +" )) and not notUsedForAln+ResNotInBothPDB")
| |
| cmd.spectrum("b", 'rainbow', "((" + objSel1 + ") or (" + objSel2 +" )) and not (notUsedForAln or ResNotInBothPDB)")
| |
| ### Delete the selection of atoms not used for alignment
| |
| ### If you would like to keep this selection intact,
| |
| ### just comment "cmd.delete" line and
| |
| ### uncomment the "cmd.disable" line abowe.
| |
| cmd.disable("notUsedForAln")
| |
| cmd.delete("notUsedForAln")
| |
| cmd.disable("ResNotInBothPDB")
| |
| cmd.delete("ResNotInBothPDB")
| |
| print "\nObjects are now colored by C-alpha displacement deviation."
| |
| print "Blue is minimum and red is maximum..."
| |
| print "White is those residues used in the alignment algorithm. Can be turned off in top of algorithm."
| |
| print "Black is residues that does not exist in both files..."
| |
| cmd.extend("ColorByDisplacementAll", ColorByDisplacementAll)
| |
| </source>
| |
|
| |
|
| [[Category:Script_Library]] | | [[Category:Script_Library]] |
| [[Category:Structural_Biology_Scripts]] | | [[Category:Structural_Biology_Scripts]] |
| | [[Category:Pymol-script-repo]] |