Grid mode: Difference between revisions
No edit summary |
ShyamSaladi (talk | contribs) (→Overview: grid mode is broadly available) |
||
| (9 intermediate revisions by 7 users not shown) | |||
| Line 4: | Line 4: | ||
'''Hint:''' When using grid_mode with many molecules, it's sometimes good to align their centers of mass. This puts them all squarely in the middle of their grid element. The '''alignto''' command from [[cealign]] can do this for you. | '''Hint:''' When using grid_mode with many molecules, it's sometimes good to align their centers of mass. This puts them all squarely in the middle of their grid element. The '''alignto''' command from [[cealign]] can do this for you. | ||
'''Hint:''' If you don't want to align the molecules (it's not | '''Hint:''' If you don't want to align the molecules (it's not really necessary for grid_mode) you can just center them on some coordinate. See [[COM]]. | ||
''' | '''Hint:''' For most uses, instead of using the above ways to calculating centering, you may be fine just executing the 'reset' command on the command line after running this mode to get full molecules in view of window and centered. | ||
== Syntax == | == Syntax == | ||
Latest revision as of 09:10, 30 March 2020
Overview
grid_mode partitions the screen into a grid and displays each molecule in one grid location. Each molecule rotates, zooms, etc in that grid. A possibly very useful option for PyMOL. Each grid area is assigned a grid_slot number. This allows you to assign objects to certain grids; very helpful. This can be useful when comparing homologous structures and you want to view and rotate the aligned structures side by side. This is similar to a multiple split screen view of the proteins.
Hint: When using grid_mode with many molecules, it's sometimes good to align their centers of mass. This puts them all squarely in the middle of their grid element. The alignto command from cealign can do this for you.
Hint: If you don't want to align the molecules (it's not really necessary for grid_mode) you can just center them on some coordinate. See COM.
Hint: For most uses, instead of using the above ways to calculating centering, you may be fine just executing the 'reset' command on the command line after running this mode to get full molecules in view of window and centered.
Syntax
# turn on grid mode
set grid_mode,1
# turn off grid mode
set grid_mode,0
Example
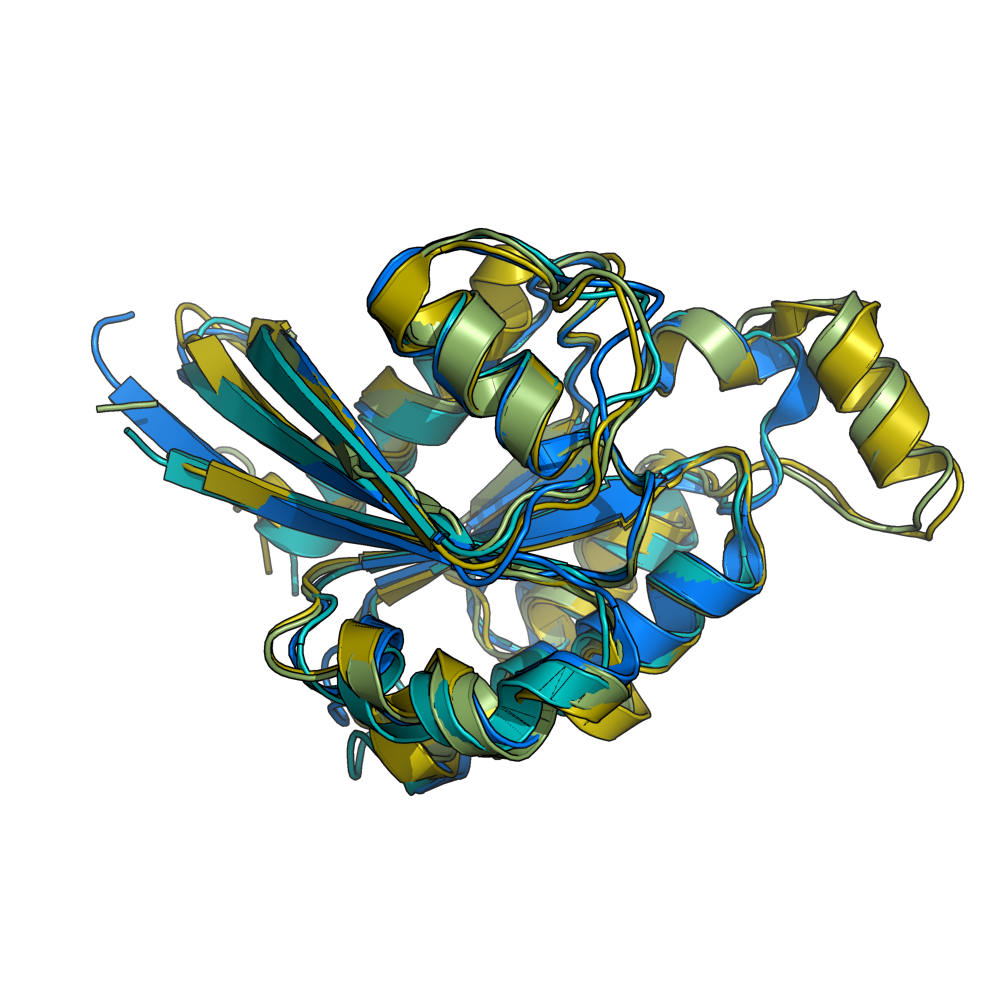
grid_mode off. Four molecules shown loaded and aligned.
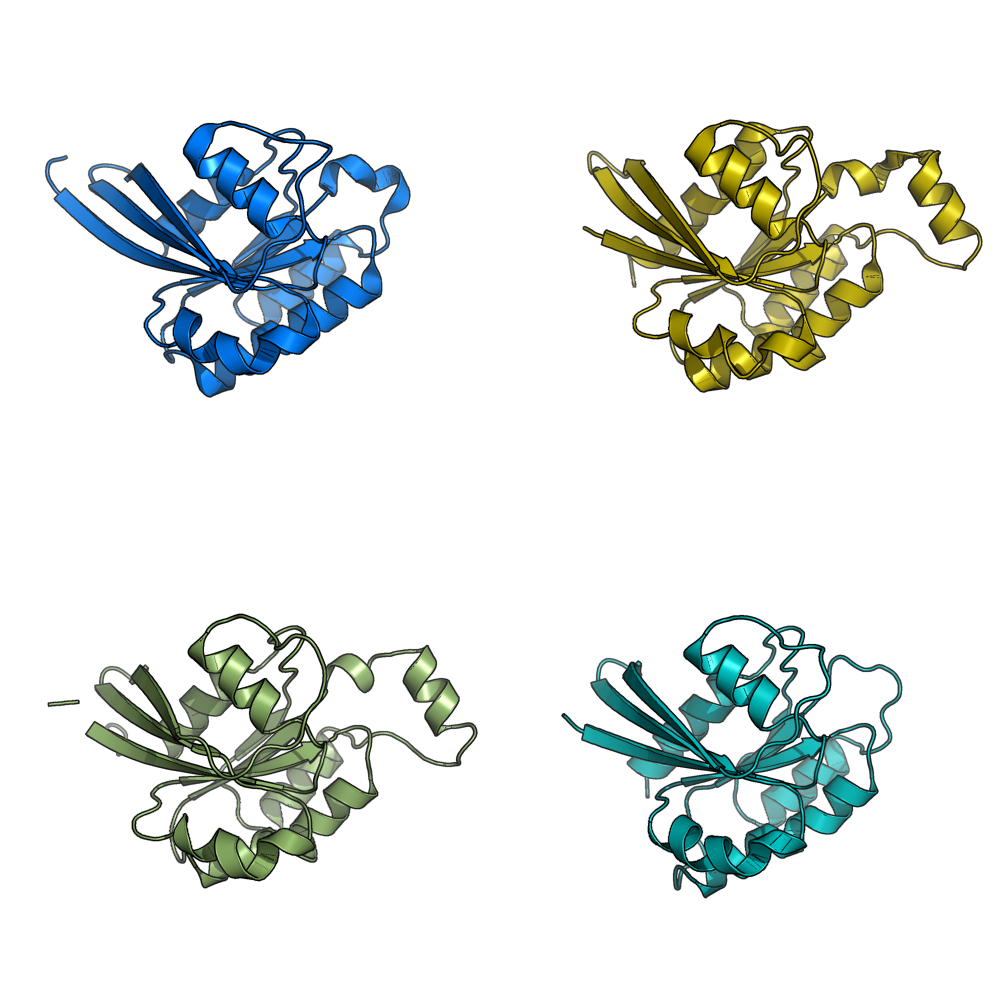
grid_mode on. Each molecule is in its own window.
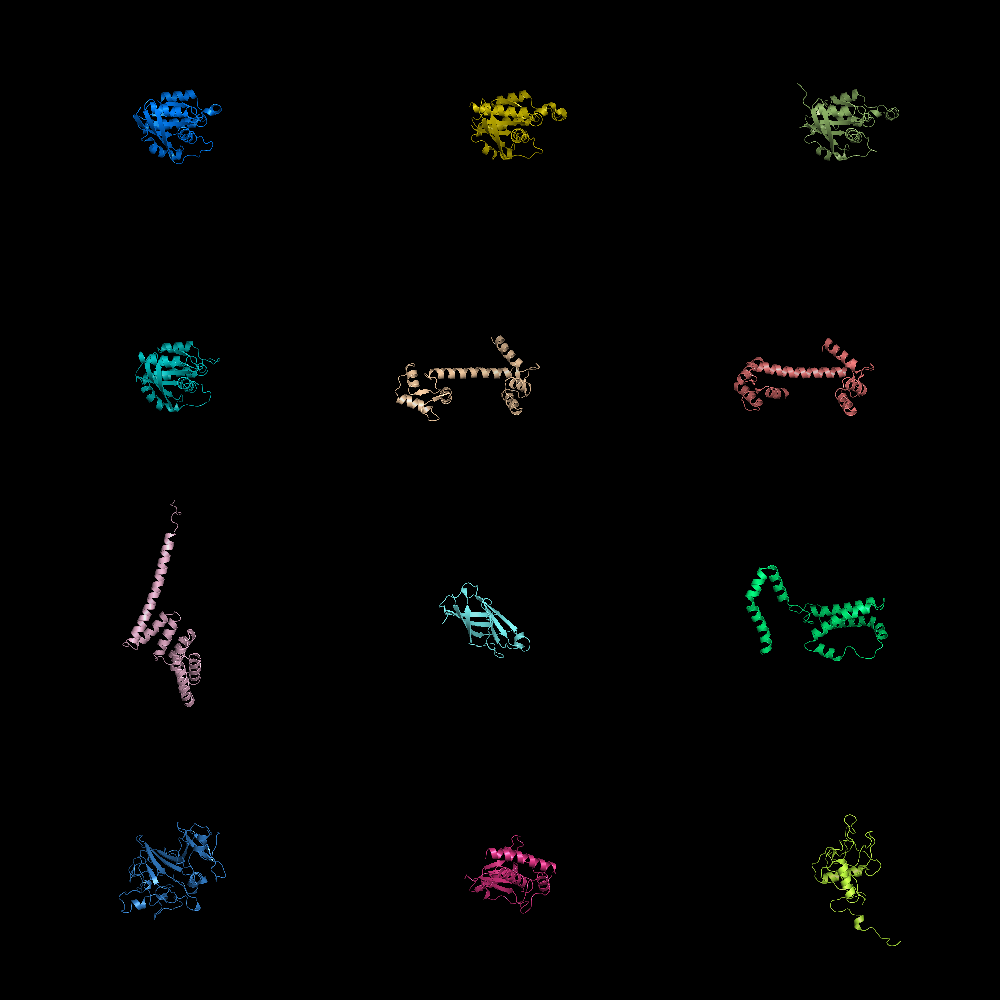
grid_mode on. Lots of molecules shown.
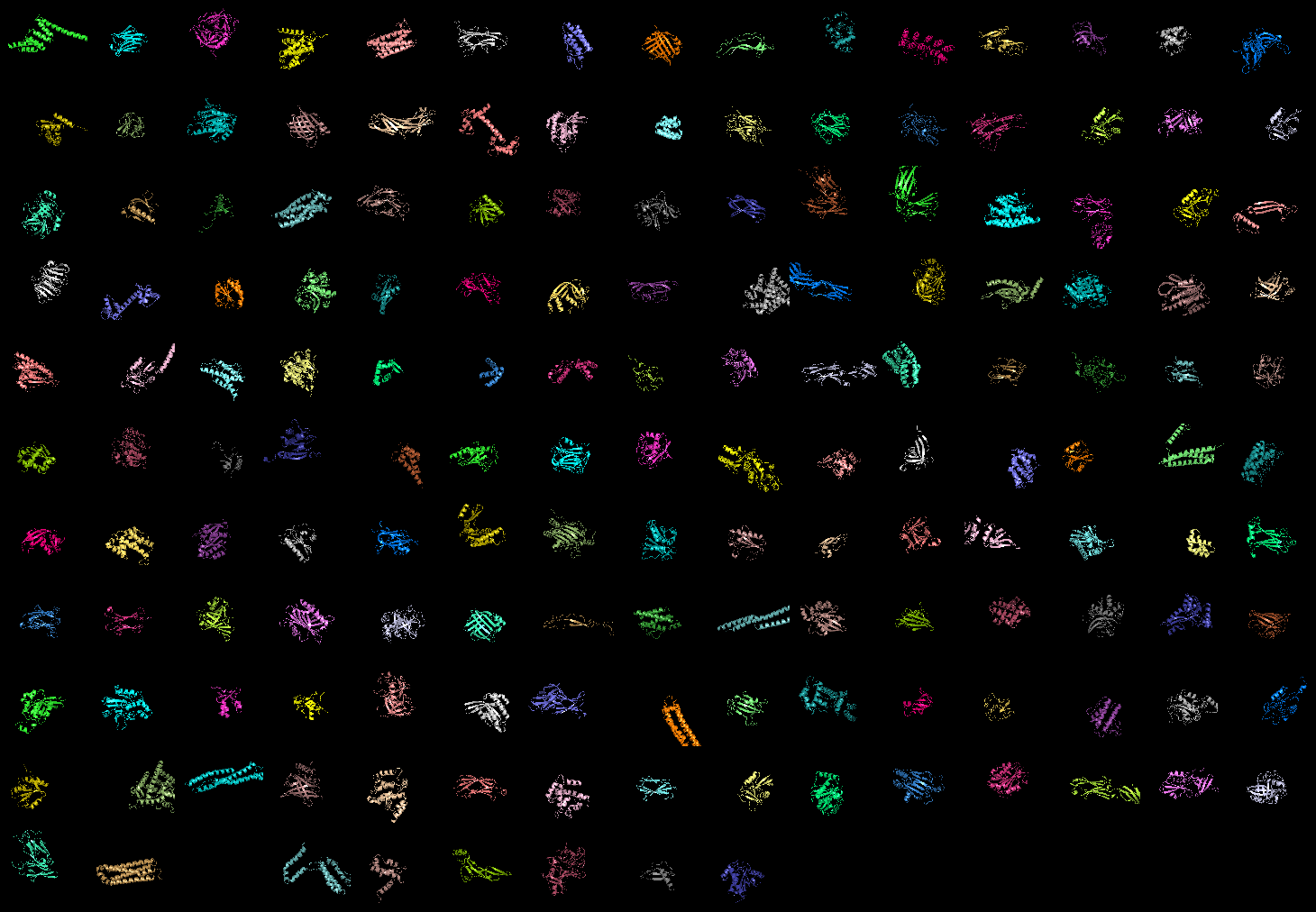
grid_mode on. 159 proteins shown!



