Cealign plugin: Difference between revisions
(cealign win32 binary. download & directions.) |
m (moved Cealign to Cealign plugin: cealign is built into PyMOL since 1.3) |
||
| (44 intermediate revisions by 8 users not shown) | |||
| Line 1: | Line 1: | ||
'''Go directly to [[Cealign#Version_0.8-RBS|DOWNLOAD]]''' | |||
Note: CEAlign is now built into PyMOL as a native command. See the open-source project page. | |||
This page is the home page of the open-source CEAlign PyMOL plugin. The CE algorithm is a fast and accurate protein structure alignment algorithm, pioneered by Drs. Shindyalov and Bourne (See | This page is the home page of the open-source CEAlign PyMOL plugin. The CE algorithm is a fast and accurate protein structure alignment algorithm, pioneered by Drs. Shindyalov and Bourne (See | ||
References | References). | ||
The source code is implemented in C with the rotations finally done by Numpy in Python. Because the computationally complex portion of the code is written in C, it's quick. That is, on my machines --- relatively fast 64-bit machines --- I can align two 400+ amino acid structures in about 0.300 s with the C++ implementation. | == Introduction == | ||
There are a few changes from the original CE publication (See Notes). The source code is implemented in C (and another in C++) with the rotations finally done by Numpy in Python (or C++ in version 0.9). Because the computationally complex portion of the code is written in C, it's quick. That is, on my machines --- relatively fast 64-bit machines --- I can align two 400+ amino acid structures in about 0.300 s with the C++ implementation. | |||
This plugs into PyMol very easily. See [[Cealign#The_Code|the code]] and [[Cealign#Examples|examples]] for installation and usage. | This plugs into PyMol very easily. See [[Cealign#The_Code|the code]] and [[Cealign#Examples|examples]] for installation and usage. | ||
| Line 13: | Line 14: | ||
'''Why should you use this?''' | '''Why should you use this?''' | ||
PyMOL's structure alignment algorithm is fast and robust. However, its first step is to perform a sequence alignment of the two selections. Thus, proteins in the '''twilight zone''' or those having a low sequence identity, may not align well. Because CE is a structure-based alignment, this is not a problem. Consider the following example. The | PyMOL's structure alignment algorithm is fast and robust. However, its first step is to perform a sequence alignment of the two selections. Thus, proteins in the '''twilight zone''' or those having a low sequence identity, may not align well. Because CE is a structure-based alignment, this is not a problem. Consider the following example. The two images below demonstrate the difference superimposing [http://www.rcsb.org/pdb/explore/explore.do?structureId=1C0m 1C0M] chain B onto [http://www.rcsb.org/pdb/explore/explore.do?structureId=1BCO 1BCO]. The first image below shows the results from PyMol's `align` command: an alignment of '''221 atoms''' (not residues) to an RMSD of '''15.7 Angstroms'''. The second image is the result of CEAlign, which used alpha carbons of '''152 residues''' with an RMSD of '''4.96 Angstroms'''. | ||
<gallery> | <gallery> | ||
Image:pymol_align.png|PyMol's results (221 atoms; 15.7 Ang. ) | |||
Image:cealign_ex1.png|Cealign's results (152 aligned; 4.96 Ang.) | Image:cealign_ex1.png|Cealign's results (152 aligned; 4.96 Ang.) | ||
</gallery> | </gallery> | ||
=== Fit vs. optAlign === | |||
====Take Home messages==== | |||
* [[fit]] and [[optAlign]] perform nearly equally as well | |||
* if you need an algorithm with an appropriate reference, use [[optAlign]] (references at bottom of page). | |||
* [[fit]] is faster -- if you're aligning many structures, use it over [[optAlign]] | |||
====Discussion==== | |||
[[optAlign]] is a function within the [[Cealign]] package that performs the optimal superposition of two objects of equal length. [[optAlign]] follows the Kabsch algorithm which is a closed form, and provably optimal solution to the problem. [[fit]] on the other hand uses the Jacobi rotations to iteratively arrive at the solution of optimal superposition. The difference in error between [[optAilgn]] and [[fit]] seems to be a non-issue (see below) as they both arrive at equivalent solutions for the rotation matrix. The two algorithms are undertake different approaches to orthogonally diagonalizing the correlation matrix. | |||
PyMOL's [[fit]] is fast and works well. If you have to use something with a known reference then check out the "optAlign" function from the qkabsch.py file that comes with this [[Cealign]] package. If not, you can just use [[fit]] and avoid installing new software. :-) | |||
optAlign is slower than fit. I just tested both on a sample NMR ensemble; and, while not an extensive validation of "fit" it shows that (1) fit is faster; and (2) fit gets the same exact RMSD as "optAlign" (when optAlign is told to use all atoms, not just CA). To make optAlign use all atoms and not just the alpha-carbon backbones, comment out (that is, put a "#" at the start of) lines 183 and 184 in qkabsch.py, where it says "CUT HERE." | |||
<source lang="python"> | |||
fetch 1nmr | |||
split_states 1nmr | |||
delete 1nmr | |||
# compare fit and optAlign RMSDs | |||
for x in cmd.get_names(): print cmd.fit("1nmr_0001", x) | |||
for x in cmd.get_names(): optAlign(x, "1nmr_0001") | |||
</source> | |||
<source lang="bash"> | |||
# results from fit | |||
0.0 | |||
4.50344991684 | |||
5.33588504791 | |||
5.78613853455 | |||
7.25597000122 | |||
6.67145586014 | |||
3.25131297112 | |||
3.36766290665 | |||
6.74802017212 | |||
5.1579709053 | |||
5.96959495544 | |||
6.68093347549 | |||
4.13217163086 | |||
5.51539039612 | |||
6.24266338348 | |||
6.03838825226 | |||
5.01363992691 | |||
5.33336305618 | |||
6.87617444992 | |||
7.797062397 | |||
#results from optAlign | |||
RMSD=0.000000 | |||
RMSD=4.503450 | |||
RMSD=5.335886 | |||
RMSD=5.786138 | |||
RMSD=7.255970 | |||
RMSD=6.671456 | |||
RMSD=3.251313 | |||
RMSD=3.367663 | |||
RMSD=6.748021 | |||
RMSD=5.157971 | |||
RMSD=5.969595 | |||
RMSD=6.680934 | |||
RMSD=4.132172 | |||
RMSD=5.515390 | |||
RMSD=6.242664 | |||
RMSD=6.038388 | |||
RMSD=5.013640 | |||
RMSD=5.333363 | |||
RMSD=6.876174 | |||
RMSD=7.797062 | |||
</source> | |||
== Examples == | == Examples == | ||
| Line 26: | Line 95: | ||
CEAlign has the semantic, and syntactic formalism of | CEAlign has the semantic, and syntactic formalism of | ||
<source lang="python"> | <source lang="python"> | ||
cealign MASTER, TARGET | |||
</source> | </source> | ||
where a post-condition of the algorithm is that the coordinates of the '''MASTER''' protein are unchanged. This allows for easier multi-protein alignments. For example, | where a post-condition of the algorithm is that the coordinates of the '''MASTER''' protein are unchanged. This allows for easier multi-protein alignments. For example, | ||
<source lang="python"> | <source lang="python"> | ||
cealign 1AUE, 1BZ4 | |||
cealign 1AUE, 1B68 | |||
cealign 1AUE, 1A7V | |||
cealign 1AUE, 1CPR | |||
</source> | </source> | ||
will superimpose all the TARGETS onto the MASTER. | will superimpose all the TARGETS onto the MASTER. | ||
| Line 51: | Line 120: | ||
to align all your proteins in PyMOL to the one called, '''PROT'''. | to align all your proteins in PyMOL to the one called, '''PROT'''. | ||
=== Results === | |||
See '''Changes''' for updates. But, overall, the results here are great. | |||
* Note: PyMOL v1.5.0.4 (svn revision 4001) has updates that improve some alignments slightly. These improved results are shown here. | |||
<gallery> | <gallery> | ||
Image:v7_1fao_1eaz.png|EASY: 1FAO vs. 1EAZ; 96 residues, 1. | Image:v7_1fao_1eaz.png|EASY: 1FAO vs. 1EAZ; 96 residues, 1.09 Ang | ||
Image:v7_1cbs_1hmt.png|EASY: 1CBS vs. 1HMT; 128 residues, 2.01 Ang | Image:v7_1cbs_1hmt.png|EASY: 1CBS vs. 1HMT; 128 residues, 2.01 Ang | ||
Image:v7_1a15_1b50.png|MODERATE: 1A15 vs 1B50; 56 residues, 2.54 Ang. | Image:v7_1a15_1b50.png|MODERATE: 1A15 vs 1B50; 56 residues, 2.54 Ang. | ||
Image:v7_1oan_1s6n.png|EASY: 1OAN vs. 1S6N (state 1); 96 residues aligned to | Image:v7_1oan_1s6n.png|EASY: 1OAN vs. 1S6N (state 1); 96 residues aligned to 2.66 Ang. RMSD. | ||
Image:v7_1rlw_1byn.png|HARD: 1RLW to 1BYN; 104 residues; 2.21 Ang. | Image:v7_1rlw_1byn.png|HARD: 1RLW to 1BYN; 104 residues; 2.21 Ang. | ||
Image:v7_1ten_3hhr.png|HARD: 1TEN vs. 3HHR; 80 residues, 2. | Image:v7_1ten_3hhr.png|HARD: 1TEN vs. 3HHR; 80 residues, 2.96 Ang. | ||
Image:v7_2sim_1nsb.png|HARD: 2SIM vs. 1NSB; 272 residues, 4. | Image:v7_2sim_1nsb.png|HARD: 2SIM vs. 1NSB; 272 residues, 4.92 Ang. | ||
Image:v7_1cew_1mol.png|HARD: 1CEW vs. 1MOL; 80 residues, | Image:v7_1cew_1mol.png|HARD: 1CEW vs. 1MOL; 80 residues, 3.67 Ang. | ||
</gallery> | </gallery> | ||
== Installation == | == Installation == | ||
===Mac OS X (10.5, 10.6)=== | |||
[[Image:Cealign mac os x.png|300px|thumb|center|CEAlign running on Mac OS X (10.5)]] | |||
* Install PyMOL under fink. | |||
* Download and install cealign (download instructions below) | |||
<source lang="bash"> | |||
sudo /sw/bin/python setup.py install | |||
</source> | |||
* In PyMOL, run the two scripts needed for cealign: "cealign.py" and "qkabsch.py". These are located in the cealign directory you previously downloaded. | |||
* Voila! | |||
* Note that the above python version must match the same version that is used by PyMOL. If you are using the pre-compiled version of MacPyMOL, the above instructions won't work. | |||
* Note: if you get an error about '''-Wno-long-double''' then your gcc is mismatched. I fixed this by pointing the symbolic link ''/usr/bin/gcc'' from ''/usr/bin/gcc-4.2'' to ''/usr/bin/gcc-4.0''. Or, in code, | |||
<source lang="bash"> | |||
# These command are commented out to stop people from copy/pasting b/c | |||
# these are possibly dangerous for your system. Ensure that /usr/bin/gcc | |||
# is a symbolic link and not a real binary. If so, I used the following | |||
# to fix the -Wno-long-double error. | |||
# sudo rm /usr/bin/gcc | |||
# sudo ln -s /usr/bin/gcc-4.0 /usr/bin/gcc | |||
</source> | |||
===Windows systems=== | ===Windows systems=== | ||
This is a quick and dirty method to | ====CEAlign 0.9==== | ||
====Requirements==== | This is a Win32 build of CEAlign 0.9 [http://pymolwiki.org/index.php/Cealign#Beta_Version_0.9] | ||
=====Requirements===== | |||
* Christoph Gohlke's latest '''unofficial''' PyMol build: http://www.lfd.uci.edu/~gohlke/#pythonlibs | |||
* "Python 2.6.2 Windows installer" from python.org: http://www.python.org/download/ | |||
* '''CEAlign09Win32.zip''' from: http://users.umassmed.edu/shivender.shandilya/pymol/CEAlign09Win32.zip | |||
=====Directions===== | |||
# Download the '''CEAlign09Win32.zip''' file | |||
# Unzip the downloaded file and follow the directions as per the included README.txt | |||
# Enjoy the ''awesomeness'' that is CEAlign! | |||
====CEAlign 0.8==== | |||
This is a quick and dirty method to use CEAlign 0.8 on Win32 system with the '''official''' Pymol builds... | |||
=====Requirements===== | |||
* Latest PyMol, installed on your system | * Latest PyMol, installed on your system | ||
* Numpy for python 2.4 | * Numpy for python 2.4 -- quick download of just what's needed: http://users.umassmed.edu/shivender.shandilya/pymol/cealign08/numpy.zip | ||
* Pre-compiled ccealign.pyd python module: http://users.umassmed.edu/Shivender.Shandilya/pymol/ccealign.zip | [Note: If this file is corrupt, you may download the latest 'Numpy for Python 2.4' directly from SourceForge.net | ||
* Modified pymolrc: http://users.umassmed.edu/Shivender.Shandilya/pymol/pymolrc | * Pre-compiled ccealign.pyd python module: http://users.umassmed.edu/Shivender.Shandilya/pymol/cealign08/ccealign.zip | ||
* Modified pymolrc: http://users.umassmed.edu/Shivender.Shandilya/pymol/cealign08/pymolrc | |||
* cealign.py and qkabsch.py from the Cealign-0.8-RBS package: download below | * cealign.py and qkabsch.py from the Cealign-0.8-RBS package: download below | ||
====Directions==== | =====Directions===== | ||
# Unzip the numpy.zip file, which will give you a folder named '''numpy''' | # Unzip the numpy.zip file, which will give you a folder named '''numpy''' | ||
# Move this entire folder to: C:\Program Files\DeLano Scientific\PyMOL\modules\ (or the corresponding location on your system) | # Move this entire folder to: C:\Program Files\DeLano Scientific\PyMOL\modules\ (or the corresponding location on your system) | ||
| Line 83: | Line 185: | ||
# Move this pyd file to: C:\Program Files\DeLano Scientific\PyMOL\py24\DLLs\ (or the corresponding location on your system) | # Move this pyd file to: C:\Program Files\DeLano Scientific\PyMOL\py24\DLLs\ (or the corresponding location on your system) | ||
# Copy the downloaded '''pymolrc''' file to: C:\Program Files\DeLano Scientific\PyMOL\ (or the corresponding location on your system) | # Copy the downloaded '''pymolrc''' file to: C:\Program Files\DeLano Scientific\PyMOL\ (or the corresponding location on your system) | ||
# Extract and copy the files cealign.py and qkabsch.py from the Cealign-0.8-RBS package to: C:\Program Files\DeLano Scientific\PyMOL\py24\Lib\ (or the corresponding location on your system) | |||
# Run PyMol and load some molecules | # Run PyMol and load some molecules | ||
# Run this command in Pymol: '''cealign molecule1, molecule2''' | # Run this command in Pymol: '''cealign molecule1, molecule2''' | ||
# Enjoy! | # Enjoy! | ||
===Gentoo Linux=== | |||
Add the science overlay via | |||
layman -a sci | |||
and emerge the cealign plugin | |||
emerge pymol-plugins-cealign | |||
===*nix systems=== | ===*nix systems=== | ||
| Line 147: | Line 256: | ||
=== Version 0.8-RBS === | === Version 0.8-RBS === | ||
* | * '''Download: [[Media:Cealign-0.8-RBS.tar.bz2|CE Align v0.8-RBS]] (bz2)''' | ||
* | * '''Download: [[Media:Cealign-0.8-RBS.zip|CE Align v0.8-RBS]] (zip)''' | ||
=== Beta Version 0.9 === | |||
Use at your own peril. Please report any problems or inconsistent alignments to this discussion page, or to me directly; my email address all over this page. | |||
'''Improvements/Changes''': | |||
* All C++ | |||
** So, faster | |||
** comes with the dependencies built in | |||
* No numpy | |||
''' Download: [[Media:Cealign-0.9.zip|CE Align v0.9]] (zip)''' | |||
== Coming Soon == | == Coming Soon == | ||
| Line 157: | Line 277: | ||
== Updates == | == Updates == | ||
=== 2008-03-25 === | |||
Pure C++ code released. See the beta version above. | |||
=== 2007-04-14 === | === 2007-04-14 === | ||
v0.8-RBS source updated. Found the bug that had been plaguing 32-bit machines. This should be the last release for a little while. | v0.8-RBS source updated. Found the bug that had been plaguing 32-bit machines. This should be the last release for a little while. | ||
Also, I provide the option of aligning based solely upon RMSD or upon the better CE-Score. See the '''References''' for information on the '''CE Score'''. | Also, I provide the option of aligning based solely upon RMSD or upon the better CE-Score. See the '''References''' for information on the '''CE Score'''. | ||
| Line 249: | Line 362: | ||
'''Solution''': This problem occurs under [http://www.apple.com/macosx Apple Mac OS X] if (a) the Apple's python executable on your machine (/usr/bin/python, currently version 2.3.5) is superseded by [http://fink.sourceforge.net/ Fink]'s python executable (/sw/bin/python, currently version 2.5) and (b) you are using [http://delsci.com/rel/099/#MacOSX precompiled versions of PyMOL] (MacPyMOL, PyMOLX11Hybrid or PyMOL for Mac OS X/X11). These executables ignore Fink's python and instead use Apple's - so, in order to run CE Align, one must install NumPy (as well as CE Align itself) using Apple's python. To do so, first download the [http://sourceforge.net/project/showfiles.php?group_id=1369&package_id=175103 Numpy source code archive] (currently version 1.0.1), unpack it, change directory to numpy-1.0.1 and specify the full path to Apple's python executable during installation: <tt>sudo /usr/bin/python setup.py install | tee install.log</tt>. Then, donwload the [http://www.pymolwiki.org/index.php/Cealign#The_Code CE Align source code archive] (currently version 0.2), unpack it, change directory to cealign-0.2 and finally install CE Align as follows: <tt>sudo /usr/bin/python setup.py install | tee install.log</tt>. | '''Solution''': This problem occurs under [http://www.apple.com/macosx Apple Mac OS X] if (a) the Apple's python executable on your machine (/usr/bin/python, currently version 2.3.5) is superseded by [http://fink.sourceforge.net/ Fink]'s python executable (/sw/bin/python, currently version 2.5) and (b) you are using [http://delsci.com/rel/099/#MacOSX precompiled versions of PyMOL] (MacPyMOL, PyMOLX11Hybrid or PyMOL for Mac OS X/X11). These executables ignore Fink's python and instead use Apple's - so, in order to run CE Align, one must install NumPy (as well as CE Align itself) using Apple's python. To do so, first download the [http://sourceforge.net/project/showfiles.php?group_id=1369&package_id=175103 Numpy source code archive] (currently version 1.0.1), unpack it, change directory to numpy-1.0.1 and specify the full path to Apple's python executable during installation: <tt>sudo /usr/bin/python setup.py install | tee install.log</tt>. Then, donwload the [http://www.pymolwiki.org/index.php/Cealign#The_Code CE Align source code archive] (currently version 0.2), unpack it, change directory to cealign-0.2 and finally install CE Align as follows: <tt>sudo /usr/bin/python setup.py install | tee install.log</tt>. | ||
[[User:Lucajovine|Luca Jovine]] 05:11, 25 January 2007 (CST). | [[User:Lucajovine|Luca Jovine]] 05:11, 25 January 2007 (CST). | ||
=== The Function SimpAlign() is not found === | |||
'''Problem''': Running CE Align gives the following error message: | |||
<source lang="python"> | |||
PyMOL>cealign 1CLL,1GGZ | |||
Traceback (most recent call last): | |||
File "C:\Program Files (x86)\DeLano Scientific\PyMOL/modules\pymol\parser.py", line 203, in parse | |||
result=apply(kw[nest][0],args[nest],kw_args[nest]) | |||
File "py24/Lib/cealign.py", line 177, in cealign | |||
curScore = simpAlign( matA, matB, mol1, mol2, stored.mol1, stored.mol2, align=0, L=len(matA) ) | |||
NameError: global name 'simpAlign' is not defined | |||
</source> | |||
I am running PyMOL v. 0.99rc6 on Win XP Professional x64 edition version 2003 sp2 and have followed the windows install procedure as described above. | |||
'''Answer''': This simply means that PyMOL couldn't find the simplAlign function. To let PyMOL know about this, you must run the following commands before running [[cealign]]: | |||
<source lang="python"> | |||
run /your/path/to/cealign/qkabsch.py | |||
run /your/path/to/cealign/cealign.py | |||
</source> | |||
but most people that use cealign would just put these two lines in their '''.pymolrc''' file. | |||
=== Short Alignments Don't Work === | |||
If you are trying to align fewer than 16 residues then use [[align]], [[super]], or [[optAlign]]. CE uses a window size of 8; and to build a path of more than one window, you need 2*8=16 residues. I will insert some code to re-route small alignments to one of the aforementioned alignment algorithms. | |||
=== It Worked A Second Ago! === | |||
[[Image:Rewind.png|thumb|right|Showing the rewind button to rewind to state 1.]] | |||
If you were using cealign (or alignto) and now the commands don't work -- that is, they return an RMSD, but don't actually superimpose the objects, then you have a simple problem dealing with states. Most likely the cause of this oddness was (1) when you issued "cealign prot1, prot2" one of them was actually an ensemble of states or (2) you are trying to align to proteins with only one state, but are not looking at state one (because the last protein you were considering had more than one state and you quit editing that protein on a state that's not state 1). To fix this, use the rewind button to get the proteins back into state 1 & reissue the cealign/alignto command. | |||
=== file is not of required architecture === | |||
This error happens on a Mac when you compile one bit of code with gcc-4.0/g++-4.0 and then try to make a library with code compiled from gcc-4.2/g++-4.2. If you recent installed Snow Leopard (Mac OS X 10.6) then this might bother you when you try to install Cealign or even PyMOL. To get around this, ensure that you're building all components with the same gcc/g++ executable. Here's how I did it, | |||
<source lang="bash"> | |||
# sudo rm /usr/bin/gcc /usr/bin/g++ | |||
# sudo ln -s /usr/bin/gcc-4.0 /usr/bin/gcc | |||
# sudo ln -s /usr/bin/g++-4.0 /usr/bin/g++ | |||
</source> | |||
I commented out those lines to stop people from blindly copy/pasting possible harmful lines. Please ensure that your /usr/bin/gcc and /usr/bin/g++ are actually symbolic links, otherwise you could be doing bad things to your computer. In my case, I only relinked gcc and not g++, hence the error. | |||
== References == | == References == | ||
Latest revision as of 16:19, 29 September 2014
Go directly to DOWNLOAD
Note: CEAlign is now built into PyMOL as a native command. See the open-source project page.
This page is the home page of the open-source CEAlign PyMOL plugin. The CE algorithm is a fast and accurate protein structure alignment algorithm, pioneered by Drs. Shindyalov and Bourne (See References).
Introduction
There are a few changes from the original CE publication (See Notes). The source code is implemented in C (and another in C++) with the rotations finally done by Numpy in Python (or C++ in version 0.9). Because the computationally complex portion of the code is written in C, it's quick. That is, on my machines --- relatively fast 64-bit machines --- I can align two 400+ amino acid structures in about 0.300 s with the C++ implementation.
This plugs into PyMol very easily. See the code and examples for installation and usage.
Comparison to PyMol
Why should you use this?
PyMOL's structure alignment algorithm is fast and robust. However, its first step is to perform a sequence alignment of the two selections. Thus, proteins in the twilight zone or those having a low sequence identity, may not align well. Because CE is a structure-based alignment, this is not a problem. Consider the following example. The two images below demonstrate the difference superimposing 1C0M chain B onto 1BCO. The first image below shows the results from PyMol's `align` command: an alignment of 221 atoms (not residues) to an RMSD of 15.7 Angstroms. The second image is the result of CEAlign, which used alpha carbons of 152 residues with an RMSD of 4.96 Angstroms.
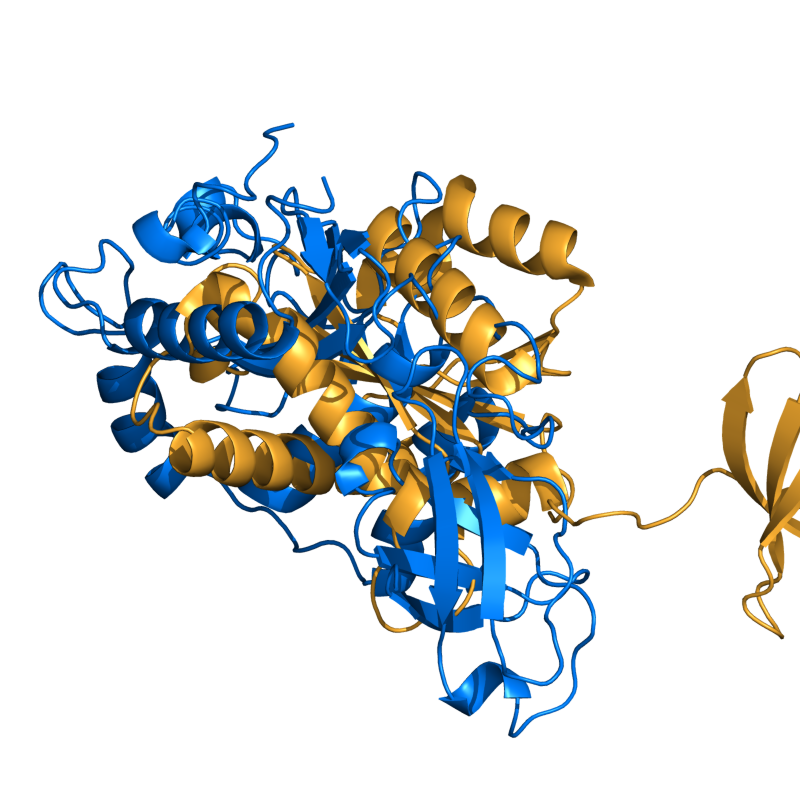
PyMol's results (221 atoms; 15.7 Ang. )
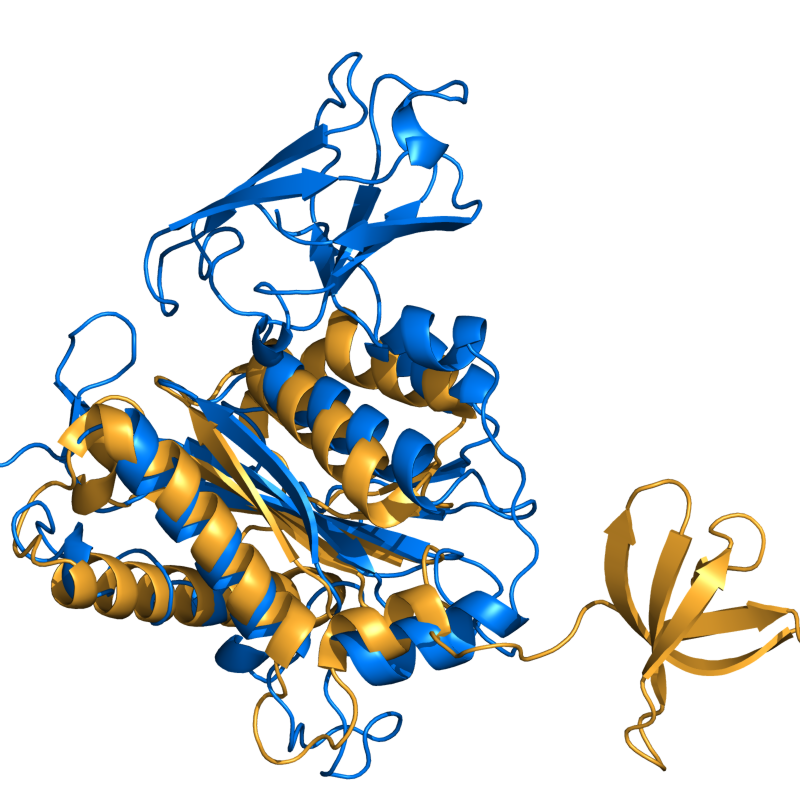
Cealign's results (152 aligned; 4.96 Ang.)


Fit vs. optAlign
Take Home messages
- fit and optAlign perform nearly equally as well
- if you need an algorithm with an appropriate reference, use optAlign (references at bottom of page).
- fit is faster -- if you're aligning many structures, use it over optAlign
Discussion
optAlign is a function within the Cealign package that performs the optimal superposition of two objects of equal length. optAlign follows the Kabsch algorithm which is a closed form, and provably optimal solution to the problem. fit on the other hand uses the Jacobi rotations to iteratively arrive at the solution of optimal superposition. The difference in error between optAilgn and fit seems to be a non-issue (see below) as they both arrive at equivalent solutions for the rotation matrix. The two algorithms are undertake different approaches to orthogonally diagonalizing the correlation matrix.
PyMOL's fit is fast and works well. If you have to use something with a known reference then check out the "optAlign" function from the qkabsch.py file that comes with this Cealign package. If not, you can just use fit and avoid installing new software. :-)
optAlign is slower than fit. I just tested both on a sample NMR ensemble; and, while not an extensive validation of "fit" it shows that (1) fit is faster; and (2) fit gets the same exact RMSD as "optAlign" (when optAlign is told to use all atoms, not just CA). To make optAlign use all atoms and not just the alpha-carbon backbones, comment out (that is, put a "#" at the start of) lines 183 and 184 in qkabsch.py, where it says "CUT HERE."
fetch 1nmr
split_states 1nmr
delete 1nmr
# compare fit and optAlign RMSDs
for x in cmd.get_names(): print cmd.fit("1nmr_0001", x)
for x in cmd.get_names(): optAlign(x, "1nmr_0001")
# results from fit
0.0
4.50344991684
5.33588504791
5.78613853455
7.25597000122
6.67145586014
3.25131297112
3.36766290665
6.74802017212
5.1579709053
5.96959495544
6.68093347549
4.13217163086
5.51539039612
6.24266338348
6.03838825226
5.01363992691
5.33336305618
6.87617444992
7.797062397
#results from optAlign
RMSD=0.000000
RMSD=4.503450
RMSD=5.335886
RMSD=5.786138
RMSD=7.255970
RMSD=6.671456
RMSD=3.251313
RMSD=3.367663
RMSD=6.748021
RMSD=5.157971
RMSD=5.969595
RMSD=6.680934
RMSD=4.132172
RMSD=5.515390
RMSD=6.242664
RMSD=6.038388
RMSD=5.013640
RMSD=5.333363
RMSD=6.876174
RMSD=7.797062
Examples
Usage
Syntax
CEAlign has the semantic, and syntactic formalism of
cealign MASTER, TARGET
where a post-condition of the algorithm is that the coordinates of the MASTER protein are unchanged. This allows for easier multi-protein alignments. For example,
cealign 1AUE, 1BZ4
cealign 1AUE, 1B68
cealign 1AUE, 1A7V
cealign 1AUE, 1CPR
will superimpose all the TARGETS onto the MASTER.
Examples
cealign 1cll and i. 42-55, 1ggz and c. A
cealign 1kao, 1ctq
cealign 1fao, 1eaz
Multiple Structure Alignments
Use the alignto command, now provided with cealign. Just type,
alignto PROT
to align all your proteins in PyMOL to the one called, PROT.
Results
See Changes for updates. But, overall, the results here are great.
- Note: PyMOL v1.5.0.4 (svn revision 4001) has updates that improve some alignments slightly. These improved results are shown here.
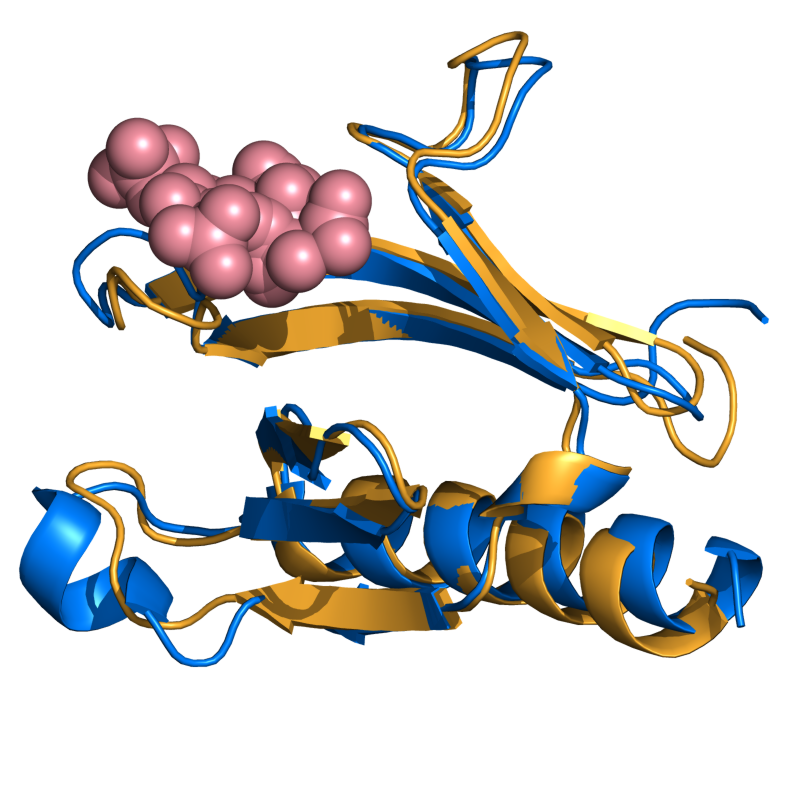
EASY: 1FAO vs. 1EAZ; 96 residues, 1.09 Ang
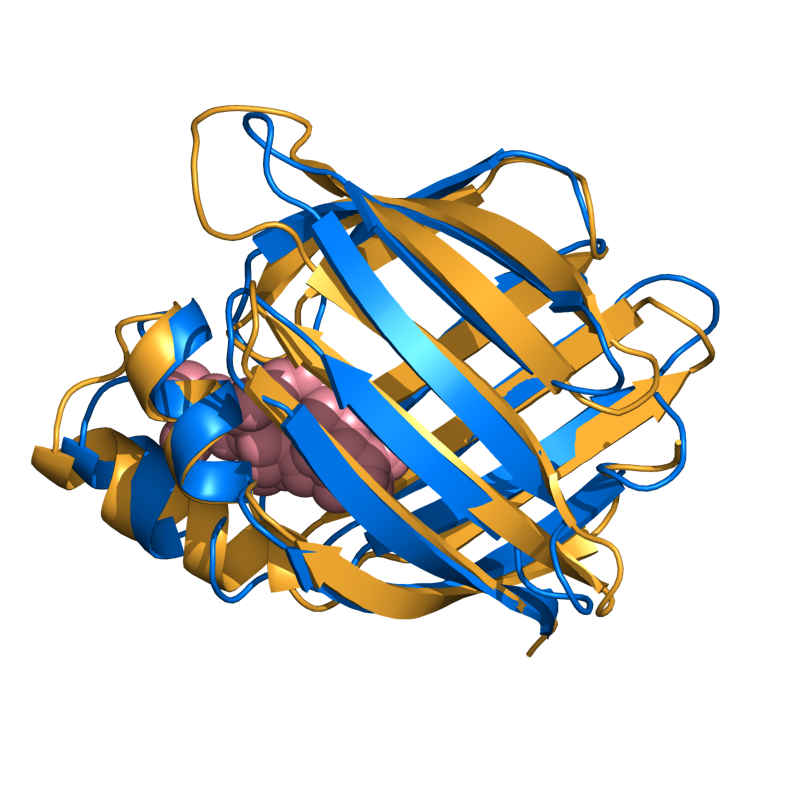
EASY: 1CBS vs. 1HMT; 128 residues, 2.01 Ang
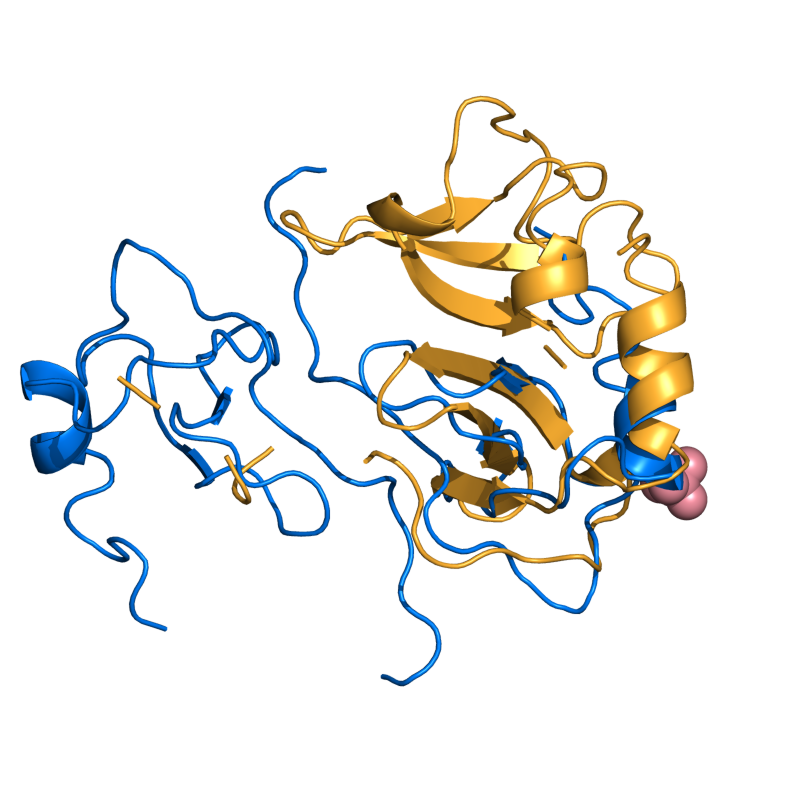
MODERATE: 1A15 vs 1B50; 56 residues, 2.54 Ang.
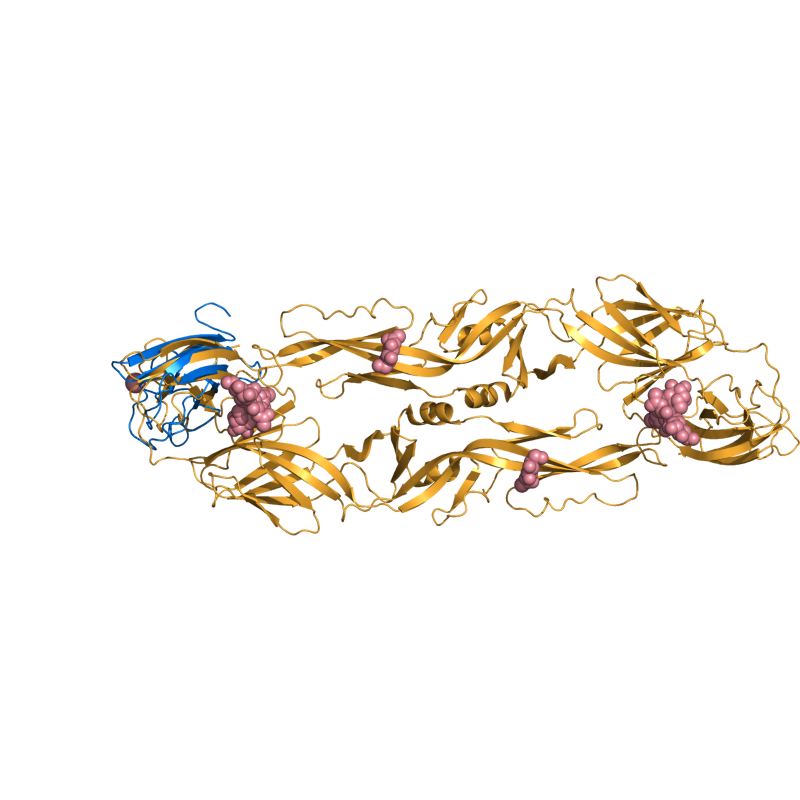
EASY: 1OAN vs. 1S6N (state 1); 96 residues aligned to 2.66 Ang. RMSD.
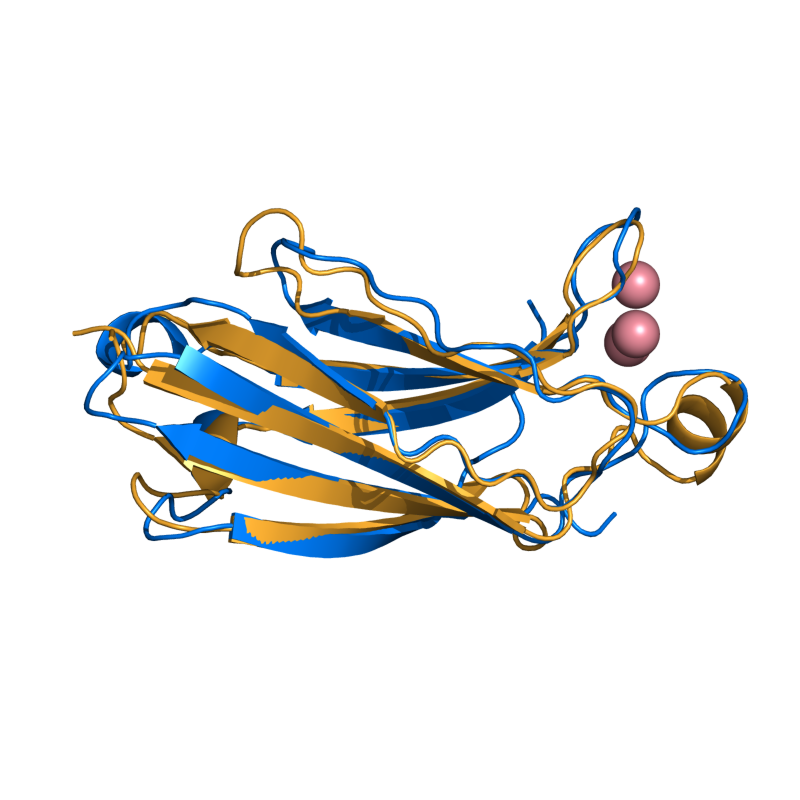
HARD: 1RLW to 1BYN; 104 residues; 2.21 Ang.
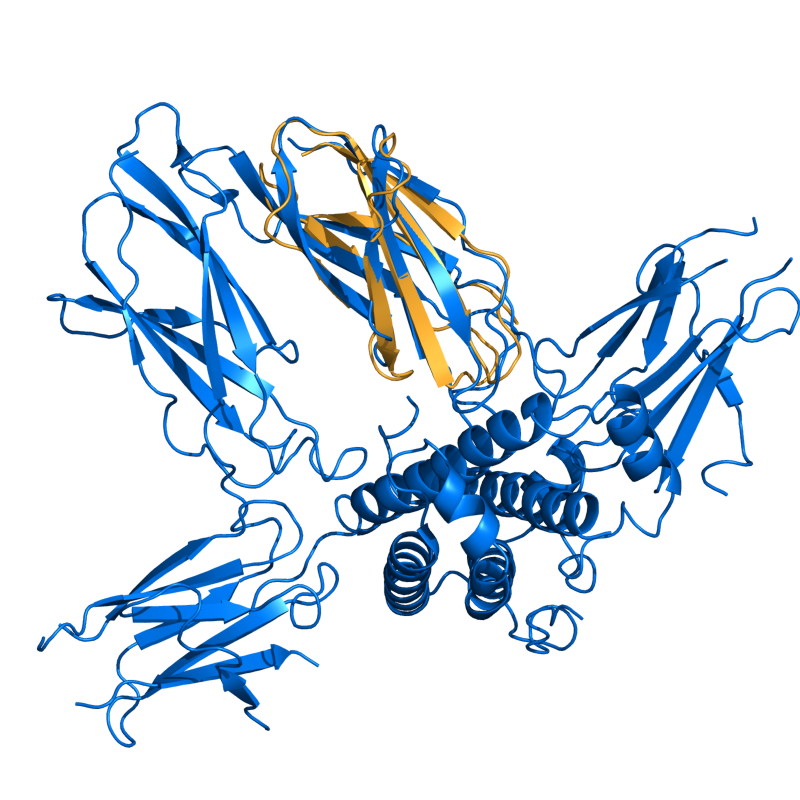
HARD: 1TEN vs. 3HHR; 80 residues, 2.96 Ang.
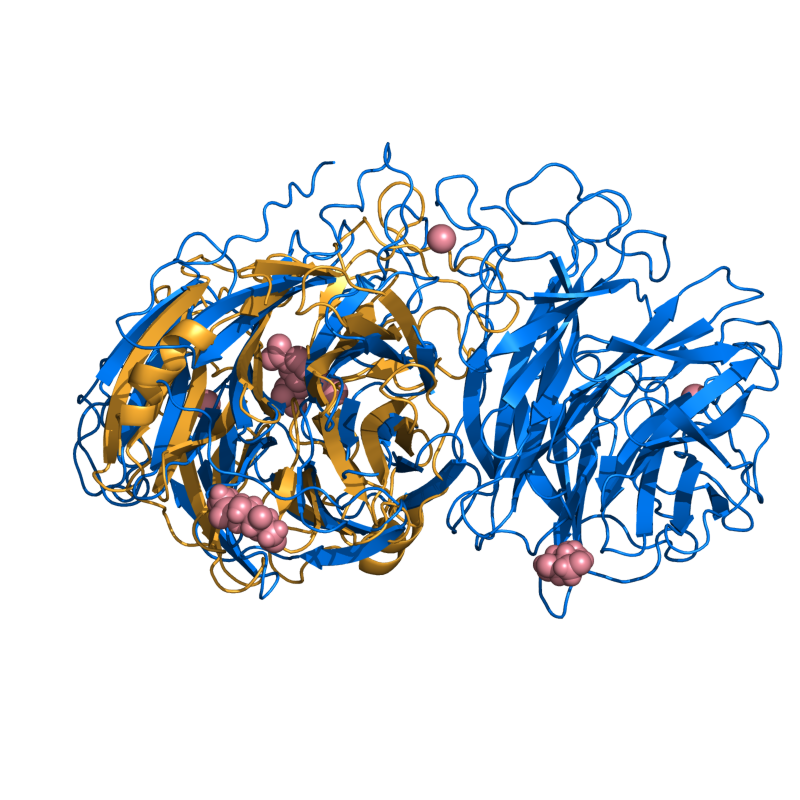
HARD: 2SIM vs. 1NSB; 272 residues, 4.92 Ang.
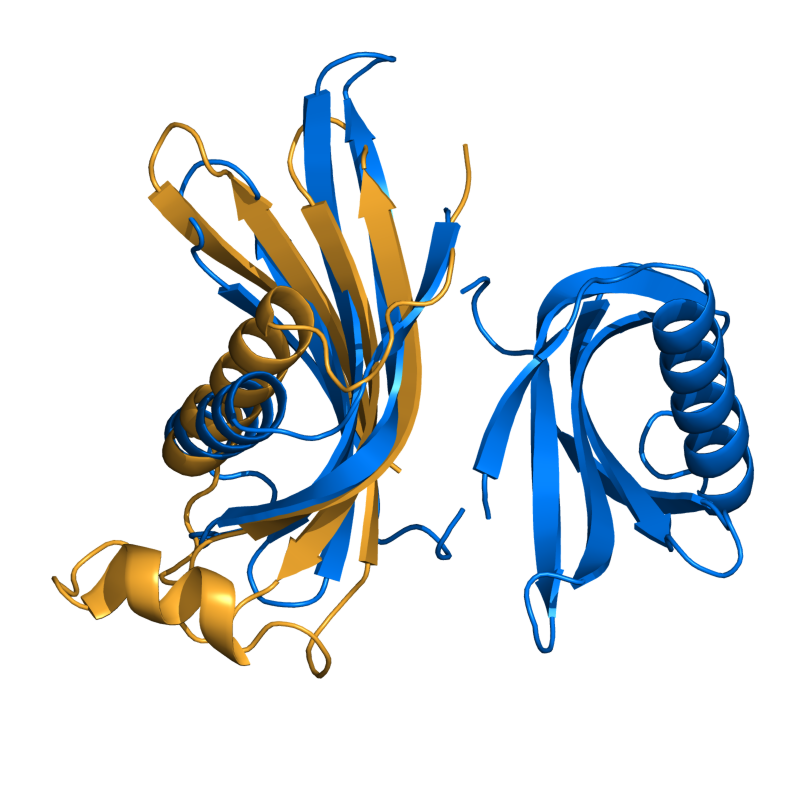
HARD: 1CEW vs. 1MOL; 80 residues, 3.67 Ang.








Installation
Mac OS X (10.5, 10.6)

- Install PyMOL under fink.
- Download and install cealign (download instructions below)
sudo /sw/bin/python setup.py install
- In PyMOL, run the two scripts needed for cealign: "cealign.py" and "qkabsch.py". These are located in the cealign directory you previously downloaded.
- Voila!
- Note that the above python version must match the same version that is used by PyMOL. If you are using the pre-compiled version of MacPyMOL, the above instructions won't work.
- Note: if you get an error about -Wno-long-double then your gcc is mismatched. I fixed this by pointing the symbolic link /usr/bin/gcc from /usr/bin/gcc-4.2 to /usr/bin/gcc-4.0. Or, in code,
# These command are commented out to stop people from copy/pasting b/c
# these are possibly dangerous for your system. Ensure that /usr/bin/gcc
# is a symbolic link and not a real binary. If so, I used the following
# to fix the -Wno-long-double error.
# sudo rm /usr/bin/gcc
# sudo ln -s /usr/bin/gcc-4.0 /usr/bin/gcc
Windows systems
CEAlign 0.9
This is a Win32 build of CEAlign 0.9 [1]
Requirements
- Christoph Gohlke's latest unofficial PyMol build: http://www.lfd.uci.edu/~gohlke/#pythonlibs
- "Python 2.6.2 Windows installer" from python.org: http://www.python.org/download/
- CEAlign09Win32.zip from: http://users.umassmed.edu/shivender.shandilya/pymol/CEAlign09Win32.zip
Directions
- Download the CEAlign09Win32.zip file
- Unzip the downloaded file and follow the directions as per the included README.txt
- Enjoy the awesomeness that is CEAlign!
CEAlign 0.8
This is a quick and dirty method to use CEAlign 0.8 on Win32 system with the official Pymol builds...
Requirements
- Latest PyMol, installed on your system
- Numpy for python 2.4 -- quick download of just what's needed: http://users.umassmed.edu/shivender.shandilya/pymol/cealign08/numpy.zip
[Note: If this file is corrupt, you may download the latest 'Numpy for Python 2.4' directly from SourceForge.net
- Pre-compiled ccealign.pyd python module: http://users.umassmed.edu/Shivender.Shandilya/pymol/cealign08/ccealign.zip
- Modified pymolrc: http://users.umassmed.edu/Shivender.Shandilya/pymol/cealign08/pymolrc
- cealign.py and qkabsch.py from the Cealign-0.8-RBS package: download below
Directions
- Unzip the numpy.zip file, which will give you a folder named numpy
- Move this entire folder to: C:\Program Files\DeLano Scientific\PyMOL\modules\ (or the corresponding location on your system)
- Unzip ccealign.zip, which will give you a file called ccealign.pyd
- Move this pyd file to: C:\Program Files\DeLano Scientific\PyMOL\py24\DLLs\ (or the corresponding location on your system)
- Copy the downloaded pymolrc file to: C:\Program Files\DeLano Scientific\PyMOL\ (or the corresponding location on your system)
- Extract and copy the files cealign.py and qkabsch.py from the Cealign-0.8-RBS package to: C:\Program Files\DeLano Scientific\PyMOL\py24\Lib\ (or the corresponding location on your system)
- Run PyMol and load some molecules
- Run this command in Pymol: cealign molecule1, molecule2
- Enjoy!
Gentoo Linux
Add the science overlay via
layman -a sci
and emerge the cealign plugin
emerge pymol-plugins-cealign
*nix systems
Requirements
- C compiler
- Python 2.4+ with distutils
- Numpy
- for User-compiled PyMOL:
python setup.py install
- for the precompiled version of PyMOL
python setup.py install --prefix "" --root /DIR_TO/pymol/ext/
- for User-compiled PyMOL:
Directions
- uncompress the distribution file cealign-VERSION.tgz
- cd cealign-VERSION
- sudo python setup.py install # if you installed by PyMOL by hand
- python setup.py install --prefix "" --root /DIR/TO/pymol/ext/ # if you are using the precompiled binary download
- insert "run DIR_TO_CEALIGN/cealign.py" and "run DIR_TO_CEALIGN/qkabsch.py" into your .pymolrc file, or just run the two Python scripts by hand.
- load some molecules
- run, cealign molecule1, molecule2
- enjoy
Pre-compiled Hackish Install
For those people that prefer to use the pre-compiled version of PyMOL, here are the basics for your install. This is a poor method of installing Cealign. I suggest users compile and install their own PyMOL. The final goal is to get
- ccealign.so module into PYMOL/ext/lib/python2.4/site-packages
- numpy installed (get the numpy directory into (or linked into) PYMOL/ext/lib/python2.4/site-packages
- and be able to run cealign.py and qkabsch.py from PyMOL.
If you can do the above three steps, cealign should run from the pre-compiled PyMOL.
In more detail, on a completely fictitious machine --- that is, I created the following commands from a fake machine and I don't expect a copy/paste of this to work anywhere, but the commands should be helpful enough to those who need it:
# NOTES:
# This is fake code: don't copy/paste it.
#
# PYMOL='dir to precompiled PyMOL install'
# CEALIGN='dir where you will unpack cealign'
# replace lib with lib64 for x86-64
# install numpy
apt-get install numpy
# link numpy to PyMOL
ln -s /usr/local/lib/python2.4/site-packages/numpy PYMOL/ext/lib/python2.4/site-packages
# download and install Cealign
wget http://www.pymolwiki.org/images/e/ed/Cealign-0.6.tar.bz2
tar -jxvf Cealign-0.6.tar.bz2
cd cealign-0.6
sudo python setup.py build
cp build/lib-XYZ-linux/ccealign.so PYMOL/ext/lib/python2.4/site-packages
# run pymol and try it out
pymol
run CEALIGN/cealign.py
run CEALIGN/qkabsch.py
fetch 1cew 1mol, async=0
cealign 1c, 1m
The Code
Please unpack and read the documentation. All comments/questions should be directed to Jason Vertrees (javertre _at_ utmb ...dot... edu).
LATEST IS v0.8-RBS. (Dedicated to Bryan Sutton for allowing me to use his computer for testing.)
Version 0.8-RBS
- Download: CE Align v0.8-RBS (bz2)
- Download: CE Align v0.8-RBS (zip)
Beta Version 0.9
Use at your own peril. Please report any problems or inconsistent alignments to this discussion page, or to me directly; my email address all over this page.
Improvements/Changes:
- All C++
- So, faster
- comes with the dependencies built in
- No numpy
Download: CE Align v0.9 (zip)
Coming Soon
- Windows binary
- Linux Binaries (32bit, x86-64)
- Better instructions for precompiled distributions
- Optimization
Updates
2008-03-25
Pure C++ code released. See the beta version above.
2007-04-14
v0.8-RBS source updated. Found the bug that had been plaguing 32-bit machines. This should be the last release for a little while.
Also, I provide the option of aligning based solely upon RMSD or upon the better CE-Score. See the References for information on the CE Score.
Troubleshooting
Post your problems/solutions here.
Unicode Issues in Python/Numpy
Problem: Running/Installing cealign gives
Traceback (most recent call last):
File "/home/byron/software/pymol_1.00b17/pymol/modules/pymol/parser.py",
line 308, in parse
File "/home/byron/software/pymol_1.00b17/pymol/modules/pymol/parsing.py",
line 410, in run_file
File "qkabsch.py", line 86, in ?
import numpy
File "/usr/lib/python2.4/site-packages/numpy/__init__.py", line 36, in ?
import core
File "/usr/lib/python2.4/site-packages/numpy/core/__init__.py", line 5, in ?
import multiarray
ImportError: /home/byron/software/pymol/ext/lib/python2.4/site-packages/numpy/core/multiarray.so:
undefined symbol: _PyUnicodeUCS4_IsWhitespace
where the important line is
undefined symbol: _PyUnicodeUCS4_IsWhitespace
This problem indicates that your Numpy Unicode is using a different byte-size for unicode characters than is the Python distribution your PyMOL is running from. For example, this can happen if you use the pre-built PyMOL and some other pre-built Numpy package.
Solution: Hand-install Numpy.
LinAlg Module Not Found
Problem: Running CE Align gives the following error message:
run qkabsch.py
Traceback (most recent call last):
File "/usr/lib/python2.4/site-packages/pymol/parser.py", line 285, in parse
parsing.run_file(exp_path(args[nest][0]),pymol_names,pymol_names)
File "/usr/lib/python2.4/site-packages/pymol/parsing.py", line 407, in run_file
execfile(file,global_ns,local_ns)
File "qkabsch.py", line 86, in ?
import numpy
File "/usr/lib/python2.4/site-packages/numpy/__init__.py", line 40, in ?
import linalg
ImportError: No module named linalg
Solution: You do not have the linear algebra module installed (or Python can't find it) on your machine. One workaround is to install Scientific Python. (on debian/ubuntu this can be done by: sudo apt-get install python-scipy) Another is to reinstall the Numpy package from source, ensuring that you have the necessary requirements for the linear algebra module (linpack, lapack, fft, etc.).
CCEAlign & NumPy Modules Not Found
Problem: Running CE Align gives the following error message:
PyMOL>run cealign.py
Traceback (most recent call last):
File "/home/local/warren/MacPyMOL060530/build/Deployment/MacPyMOL.app/pymol/modules/pymol/parser.py", line 297, in parse
File "/home/local/warren/MacPyMOL060530/build/Deployment/MacPyMOL.app/pymol/modules/pymol/parsing.py", line 408, in run_file
File "/usr/local/pymol/scripts/cealign-0.1/cealign.py", line 59, in ?
from ccealign import ccealign
ImportError: No module named ccealign
run qkabsch.py
Traceback (most recent call last):
File "/home/local/warren/MacPyMOL060530/build/Deployment/MacPyMOL.app/pymol/modules/pymol/parser.py", line 297, in parse
File "/home/local/warren/MacPyMOL060530/build/Deployment/MacPyMOL.app/pymol/modules/pymol/parsing.py", line 408, in run_file
File "qkabsch.py", line 86, in ?
import numpy
ImportError: No module named numpy
Solution: This problem occurs under Apple Mac OS X if (a) the Apple's python executable on your machine (/usr/bin/python, currently version 2.3.5) is superseded by Fink's python executable (/sw/bin/python, currently version 2.5) and (b) you are using precompiled versions of PyMOL (MacPyMOL, PyMOLX11Hybrid or PyMOL for Mac OS X/X11). These executables ignore Fink's python and instead use Apple's - so, in order to run CE Align, one must install NumPy (as well as CE Align itself) using Apple's python. To do so, first download the Numpy source code archive (currently version 1.0.1), unpack it, change directory to numpy-1.0.1 and specify the full path to Apple's python executable during installation: sudo /usr/bin/python setup.py install | tee install.log. Then, donwload the CE Align source code archive (currently version 0.2), unpack it, change directory to cealign-0.2 and finally install CE Align as follows: sudo /usr/bin/python setup.py install | tee install.log. Luca Jovine 05:11, 25 January 2007 (CST).
The Function SimpAlign() is not found
Problem: Running CE Align gives the following error message:
PyMOL>cealign 1CLL,1GGZ
Traceback (most recent call last):
File "C:\Program Files (x86)\DeLano Scientific\PyMOL/modules\pymol\parser.py", line 203, in parse
result=apply(kw[nest][0],args[nest],kw_args[nest])
File "py24/Lib/cealign.py", line 177, in cealign
curScore = simpAlign( matA, matB, mol1, mol2, stored.mol1, stored.mol2, align=0, L=len(matA) )
NameError: global name 'simpAlign' is not defined
I am running PyMOL v. 0.99rc6 on Win XP Professional x64 edition version 2003 sp2 and have followed the windows install procedure as described above.
Answer: This simply means that PyMOL couldn't find the simplAlign function. To let PyMOL know about this, you must run the following commands before running cealign:
run /your/path/to/cealign/qkabsch.py
run /your/path/to/cealign/cealign.py
but most people that use cealign would just put these two lines in their .pymolrc file.
Short Alignments Don't Work
If you are trying to align fewer than 16 residues then use align, super, or optAlign. CE uses a window size of 8; and to build a path of more than one window, you need 2*8=16 residues. I will insert some code to re-route small alignments to one of the aforementioned alignment algorithms.
It Worked A Second Ago!

If you were using cealign (or alignto) and now the commands don't work -- that is, they return an RMSD, but don't actually superimpose the objects, then you have a simple problem dealing with states. Most likely the cause of this oddness was (1) when you issued "cealign prot1, prot2" one of them was actually an ensemble of states or (2) you are trying to align to proteins with only one state, but are not looking at state one (because the last protein you were considering had more than one state and you quit editing that protein on a state that's not state 1). To fix this, use the rewind button to get the proteins back into state 1 & reissue the cealign/alignto command.
file is not of required architecture
This error happens on a Mac when you compile one bit of code with gcc-4.0/g++-4.0 and then try to make a library with code compiled from gcc-4.2/g++-4.2. If you recent installed Snow Leopard (Mac OS X 10.6) then this might bother you when you try to install Cealign or even PyMOL. To get around this, ensure that you're building all components with the same gcc/g++ executable. Here's how I did it,
# sudo rm /usr/bin/gcc /usr/bin/g++
# sudo ln -s /usr/bin/gcc-4.0 /usr/bin/gcc
# sudo ln -s /usr/bin/g++-4.0 /usr/bin/g++
I commented out those lines to stop people from blindly copy/pasting possible harmful lines. Please ensure that your /usr/bin/gcc and /usr/bin/g++ are actually symbolic links, otherwise you could be doing bad things to your computer. In my case, I only relinked gcc and not g++, hence the error.
References
Text taken from PubMed and formatted for the wiki. The first reference is the most important for this code.
- Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998 Sep;11(9):739-47. PMID: 9796821 [PubMed - indexed for MEDLINE]
- Jia Y, Dewey TG, Shindyalov IN, Bourne PE. A new scoring function and associated statistical significance for structure alignment by CE. J Comput Biol. 2004;11(5):787-99. PMID: 15700402 [PubMed - indexed for MEDLINE]
- Pekurovsky D, Shindyalov IN, Bourne PE. A case study of high-throughput biological data processing on parallel platforms. Bioinformatics. 2004 Aug 12;20(12):1940-7. Epub 2004 Mar 25. PMID: 15044237 [PubMed - indexed for MEDLINE]
- Shindyalov IN, Bourne PE. An alternative view of protein fold space. Proteins. 2000 Feb 15;38(3):247-60. PMID: 10713986 [PubMed - indexed for MEDLINE]
License
The CEAlign and all its subprograms that I wrote, are released under the open source Free BSD License (BSDL).