FindSurfaceCharge: Difference between revisions
Jump to navigation
Jump to search
TeddyWarner (talk | contribs) (Creation of new variant of pymol function) |
PedroLacerda (talk | contribs) No edit summary |
||
| (9 intermediate revisions by one other user not shown) | |||
| Line 1: | Line 1: | ||
{{Infobox script-repo | {{Infobox script-repo | ||
|type = module | |type = module | ||
|filename = | |filename = scripts/findSurfaceCharge.py | ||
|author = [[User:TeddyWarner|Teddy Warner]] | |author = [[User:TeddyWarner|Teddy Warner]] | ||
|license = BSD-2-Clause | |license = BSD-2-Clause | ||
}} | }} | ||
Drawing upon the [[findSurfaceResidues]] script, the [[findSurfaceCharge]] script will identify and output a list of all charged residues on the surface of a selectionand calculates the ionization state of a protein at a given pH. The charge can be calculated for either a folded or denatured protein. This function is intended to be used to give buffer conditions for mass spectrometry. | |||
= Usage = | = Usage = | ||
findSurfaceCharge [selection, [pH, [folded ,[cutoff]]]] | |||
= Arguments = | = Arguments = | ||
* '''selection''' = str: The object or selection for which to find exposed residues {default: all} | |||
* '''pH''' = float: The pH to calculate the surface charge at {default: 7.0} | * '''pH''' = float: The pH to calculate the surface charge at {default: 7.0} | ||
* ''' | * '''folded''' = bool: Whether the program should calculate the charge of a folded (True) or denatured (False) protein. | ||
* '''cutoff''' = float: The cutoff in square Angstroms that defines exposed or not. Those atoms with > cutoff Ang^2 exposed will be considered ''exposed'' {default: 2.5 Ang^2} | * '''cutoff''' = float: The cutoff in square Angstroms that defines exposed or not. Those atoms with > cutoff Ang^2 exposed will be considered ''exposed'' {default: 2.5 Ang^2} | ||
= Examples = | = Examples = | ||
<gallery> | <gallery> | ||
File:SurfaceCharge.PNG|Result of 4FIX.pdb at pH 7.0. | |||
</gallery> | </gallery> | ||
<source lang="python"> | <source lang="python"> | ||
run findSurfaceResiduesListCharged.py | run findSurfaceResiduesListCharged.py | ||
fetch 4FIX | fetch 4FIX | ||
findSurfaceResiduesListCharged | findSurfaceResiduesListCharged | ||
# see how pH changes the protein surface charge: | # see how pH changes the protein surface charge: | ||
findSurfaceCharge("4fix",7.0) | |||
Exposed charged residues: | |||
ERREDRKEE... | |||
The expected surface charge of 4fix at pH 7.0 is: +3.24 | |||
findSurfaceCharge("4fix",7.0,False) | |||
Charged residues: | |||
HHHHHHRHERREDRKEE... | |||
The expected charge of denatured 4fix at pH 7.0 is: +0.13 | |||
findSurfaceCharge("4fix",10.0) | |||
Charged residues: ... | |||
The expected surface charge of 4fix at pH 10.0 is: -3.86 | |||
</source> | </source> | ||
| Line 48: | Line 57: | ||
[[Category:Biochemical_Scripts]] | [[Category:Biochemical_Scripts]] | ||
[[Category:Structural_Biology_Scripts]] | [[Category:Structural_Biology_Scripts]] | ||
[[Category:Pymol-script-repo]] | |||
__NOTOC__ | __NOTOC__ | ||
Latest revision as of 23:04, 22 June 2025
| Type | Python Module |
|---|---|
| Download | scripts/findSurfaceCharge.py |
| Author(s) | Teddy Warner |
| License | BSD-2-Clause |
| This code has been put under version control in the project Pymol-script-repo | |
Drawing upon the findSurfaceResidues script, the findSurfaceCharge script will identify and output a list of all charged residues on the surface of a selectionand calculates the ionization state of a protein at a given pH. The charge can be calculated for either a folded or denatured protein. This function is intended to be used to give buffer conditions for mass spectrometry.
Usage
findSurfaceCharge [selection, [pH, [folded ,[cutoff]]]]
Arguments
- selection = str: The object or selection for which to find exposed residues {default: all}
- pH = float: The pH to calculate the surface charge at {default: 7.0}
- folded = bool: Whether the program should calculate the charge of a folded (True) or denatured (False) protein.
- cutoff = float: The cutoff in square Angstroms that defines exposed or not. Those atoms with > cutoff Ang^2 exposed will be considered exposed {default: 2.5 Ang^2}
Examples
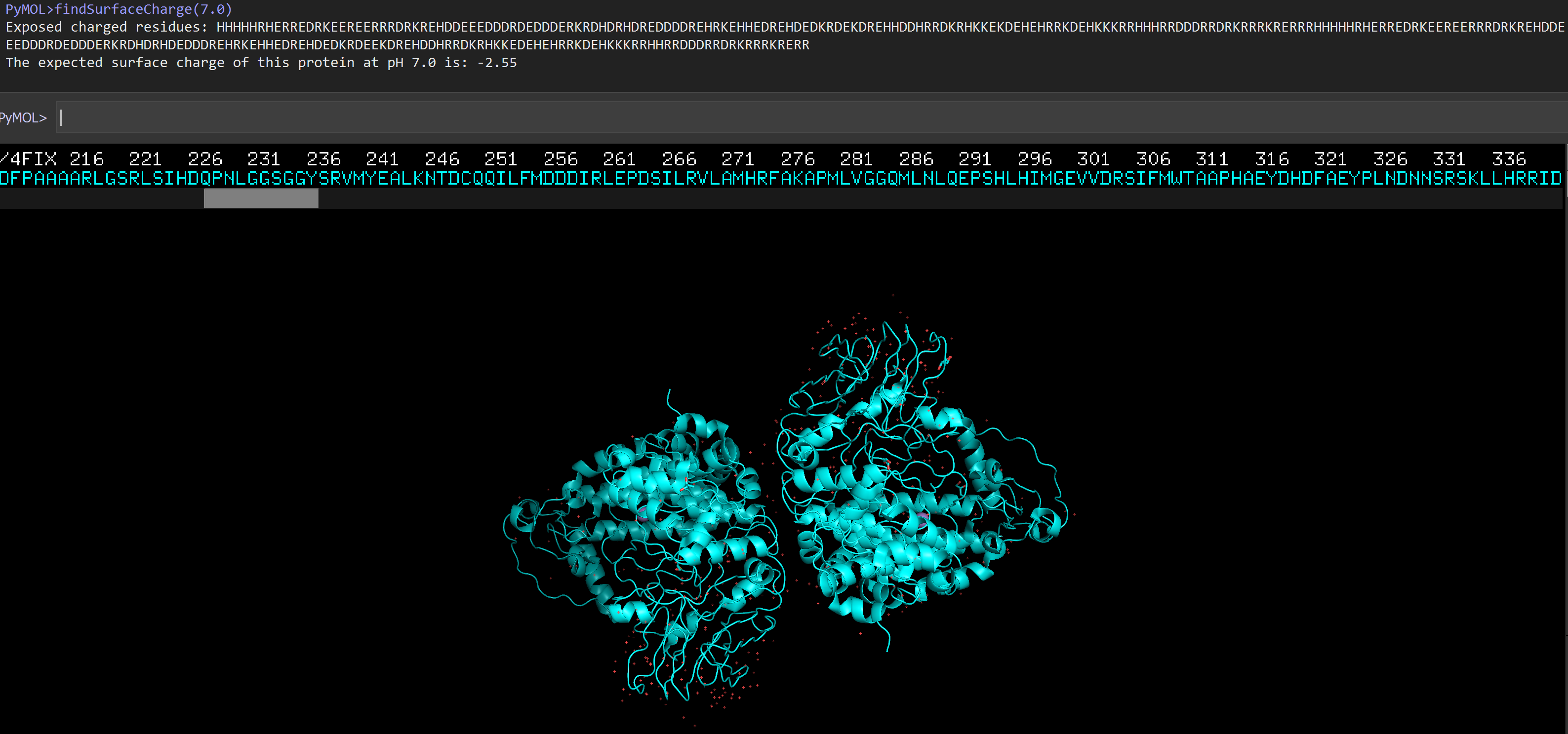
Result of 4FIX.pdb at pH 7.0.

run findSurfaceResiduesListCharged.py
fetch 4FIX
findSurfaceResiduesListCharged
# see how pH changes the protein surface charge:
findSurfaceCharge("4fix",7.0)
Exposed charged residues:
ERREDRKEE...
The expected surface charge of 4fix at pH 7.0 is: +3.24
findSurfaceCharge("4fix",7.0,False)
Charged residues:
HHHHHHRHERREDRKEE...
The expected charge of denatured 4fix at pH 7.0 is: +0.13
findSurfaceCharge("4fix",10.0)
Charged residues: ...
The expected surface charge of 4fix at pH 10.0 is: -3.86