Colorbydisplacement: Difference between revisions
(Created page with "== Acknowledgement == This pymol script is made by Troels Emtekær Linnet. This script is based on the scaffold from ColorByRMSD. Peace love and harmony goes to Shivender Sh...") |
|||
| Line 24: | Line 24: | ||
=== Examples === | === Examples === | ||
< | <syntaxhighlight lang="python"> | ||
ColorByDisplacementCA O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t | ColorByDisplacementCA O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t | ||
ColorByDisplacementAll O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t | ColorByDisplacementAll O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t | ||
| Line 33: | Line 33: | ||
ColorByDisplacementCA O5NT, C5NT, resi 26-355, resi 26-355 | ColorByDisplacementCA O5NT, C5NT, resi 26-355, resi 26-355 | ||
ColorByDisplacementAll O5NT, C5NT, resi 26-355, resi 26-355 | ColorByDisplacementAll O5NT, C5NT, resi 26-355, resi 26-355 | ||
</ | </syntaxhighlight> | ||
<gallery heights="240px" widths="340px"> | <gallery heights="240px" widths="340px"> | ||
Revision as of 10:11, 1 December 2011
Acknowledgement
This pymol script is made by Troels Emtekær Linnet.
This script is based on the scaffold from ColorByRMSD. Peace love and harmony goes to Shivender Shandilya and Jason Vertrees.
Introduction
This script allows you to color two structures by distance displacement between an Open and Closed form of a protein, as calculated by PyMol's internal distance command. The pairwise distance is calculated between, C-alpha or all-atoms. The distance displacement values are stored as B-factors of these residues, which are colored by a rainbow color spectrum, with blue specifying minimum and red indicating maximum.
Code
Do keep in mind, all original B-factors values are overwritten!
There exist two versions.
ColorByDisplacementCA is quick and is between CA atoms. Ideal for helices representation.
ColorByDisplacementAll is between All atoms in residues and is quite slow => 3-5 mins for a run. Ideal for sticks representation.
You have to specify which residues should be used in the alignment procedure, or it will take all residues as standard
V.2 is implemented the 2011.01.06 - Due to a bug in coloring.
Bug in code
A bug in the boolean operator of the spectrum command has been found. This versions work for version 1.3 Educational product.
For other versions of pymol, try to change (comment/uncomment) the cmd.spectrum line.
The other spectrum line works for Open-Source PyMOL 1.2r3pre, Incentive product
Examples
ColorByDisplacementCA O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t
ColorByDisplacementAll O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t
ColorByDisplacementCA O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t, AlignedWhite='no'
ColorByDisplacementAll O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t, AlignedWhite='no'
ColorByDisplacementCA O5NT, C5NT, resi 26-355, resi 26-355
ColorByDisplacementAll O5NT, C5NT, resi 26-355, resi 26-355

ColorByDisplacementCA used on 1HP1 and 1HPU aligned and colored by distance displacement.
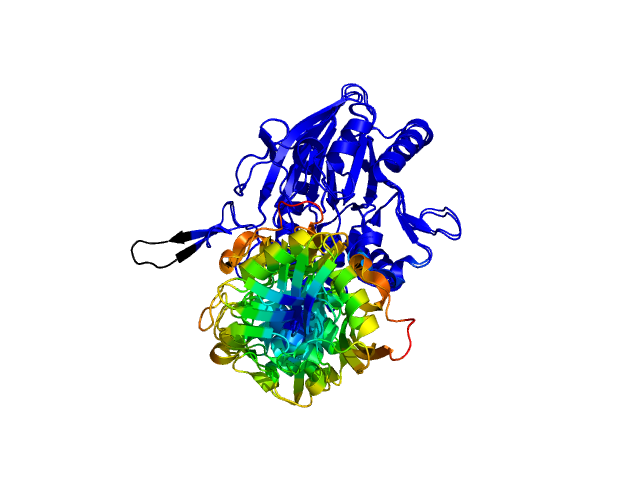
ColorByDisplacementCA used on 1HP1 and 1HPU aligned and colored by distance displacement.
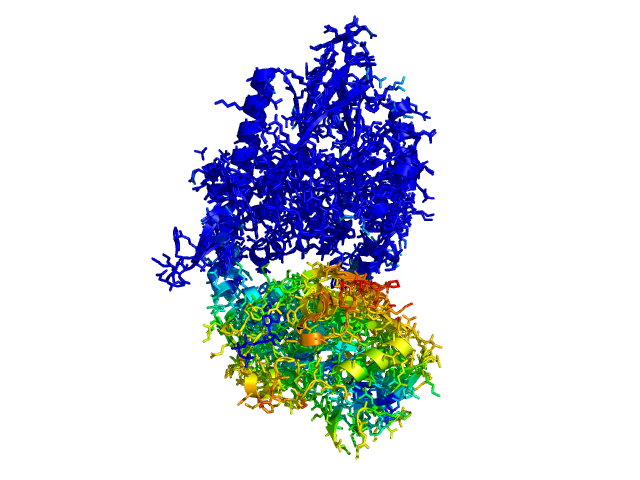
ColorByDisplacementAll used on 1HP1 and 1HPU aligned and colored by distance displacement.
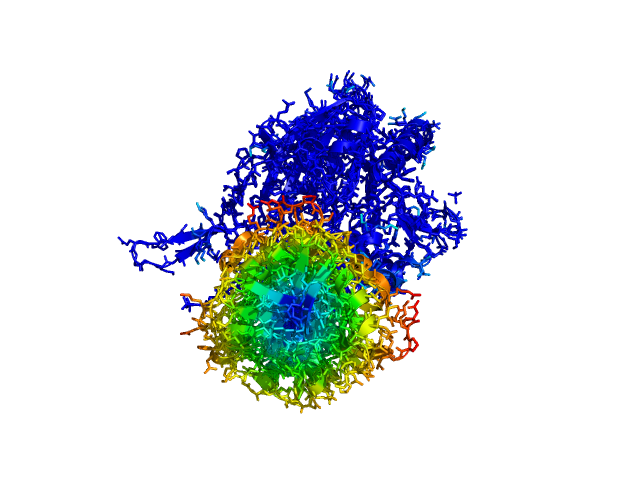
ColorByDisplacementAll used on 1HP1 and 1HPU aligned and colored by distance displacement.




Dark blue is low displacement, higher displacements are in orange/yellow/red.
Residues used for alignment is colored white. Can be turned off in top of algorithm.
Residues not in both pdb files is colored black
Example Pymol Script
cd C:\Users\tlinnet\Documents\My Dropbox\Speciale\5NT-project\Mutant-construct\Distance-Plot
#cd /homes/linnet/Documents/Speciale/5NT-project/Mutant-construct/Distance-Plot
### load pdb files and rename
load 1HP1.pdb, O5NT-1HP1
load 1HPU.pdb, C5NT-1HPU
hide everything
### Select asymmetric units from pdb file
create O5NT, /O5NT-1HP1//A
create C5NT, /C5NT-1HPU//C
delete O5NT-1HP1
delete C5NT-1HPU
cartoon auto
show cartoon, O5NT
show cartoon, C5NT
set cartoon_fancy_helices=1
set bg,[1,1,1]
set auto_zoom, off
### Make sharper
set fog=0
### Load my function, and run function with input
run ColorByDisplacement.py
ColorByDisplacementCA O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t
#ColorByDisplacementAll O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t
set_view (\
0.094686687, -0.390707940, 0.915631354,\
0.809000611, -0.505792081, -0.299485058,\
0.580131471, 0.769104064, 0.268191338,\
0.000000000, 0.000000000, -280.940521240,\
26.240486145, 46.146961212, 21.702068329,\
231.830673218, 330.050415039, -20.000000000 )
Python Code
import pymol
import cmd
from pymol import stored
### Thanks for inspiration from:
"""
--- ColorByRMSD: RMSD based coloring ---
Authors : Shivender Shandilya; Jason Vertrees
Program : ColorByRMSD
Date : July 2009
http://www.pymolwiki.org/index.php/ColorByRMSD
"""
### Author Troels Linnet - troels.linnet att bbz.uni-leipzig.de
"""
--- ColorByDisplacementCA: Displacement based coloring ---
Authors : Troels E. Linnet
Program : ColorByDisplacementCA
Date : January 2011
email: troels.linnet att bbz.uni-leipzig.de
"""
"""
ColorByDisplacementCA --
Show the distance displacement deviation in color to more easily see variable regions.
PARAMS
objSel1 (valid PyMOL object or selection)
The first object
objSel2 (valid PyMOL object or selection)
The second object
doColor (boolean, either True or False)
If doColor=True then a simple representation is created to
highlight the differences. If False, then no changes are made.
DEFAULT: False
RETURNS
None.
SIDE-EFFECTS
Modifies the B-factor columns in your original structures.
"""
def strTrue(p):
return p[0].upper() == "T"
# The main function that assigns current displacement distance as the new B-factor
def displacementUpdateB(objA, alnAri, objB, alnBri):
### If residue is unassigned in one of the pdb files, we reset its value
for x in range(len(alnAri)):
s1 = objA + " and name CA and resi " + alnAri[x]
cmd.alter( s1, "b = " + str(-0.01))
for x in range(len(alnBri)):
s2 = objB + " and name CA and resi " + alnBri[x]
cmd.alter( s2, "b = " + str(-0.01))
cmd.sort(objA); cmd.sort(objB)
for x in range(len(alnAri)):
s1 = objA + " and name CA and resi " + alnAri[x]
s2 = objB + " and name CA and resi " + alnAri[x]
### Names starting with __ (underscores) are normally hidden by PyMOL
tempObject = "__tempObject"
Displacement = cmd.distance(tempObject, s1, s2)
cmd.alter( s1, "b = " + str(Displacement))
cmd.alter( s2, "b = " + str(Displacement))
cmd.delete(tempObject)
cmd.sort(objA); cmd.sort(objB)
def ColorByDisplacementCA(objSel1, objSel2, super1='all', super2='all', doColor="True", doAlign="True", AlignedWhite='yes'):
### First create backup copies; names starting with __ (underscores) are normally hidden by PyMOL
tObj1, tObj2, aln = "__tempObj1", "__tempObj2", "__aln"
if strTrue(doAlign):
### Create temp objects
cmd.create( tObj1, objSel1 )
cmd.create( tObj2, objSel2 )
### Align and make create an object aln which indicates which atoms were paired between the two structures
### Super is must faster than align http://www.pymolwiki.org/index.php/Super
cmd.super(tObj1 + ' and ' + str(super1), tObj2 + ' and ' + str(super2), object=aln)
### Modify the original matrix of object1 from the alignment
cmd.matrix_copy(tObj1, objSel1)
else:
### Create temp objects
cmd.create( tObj1, objSel1 )
cmd.create( tObj2, objSel2 )
### Align and make create an object aln which indicates which atoms were paired between the two structures
### Super is must faster than align http://www.pymolwiki.org/index.php/Super
cmd.super(tObj1 + ' and ' + str(super1), tObj2 + ' and ' + str(super2), object=aln)
### Modify the B-factor columns of the original objects,
### in order to identify the residues NOT used for alignment, later on
cmd.alter( objSel1 + " or " + objSel2, "b=-0.2")
cmd.alter( tObj1 + " or " + tObj2, "chain='A'")
cmd.alter( tObj1 + " or " + tObj2, "segi='A'")
### Update pymol internal representations; one of these should do the trick
cmd.refresh(); cmd.rebuild(); cmd.sort(tObj1); cmd.sort(tObj2)
### Create lists for storage
stored.alnAres, stored.alnBres = [], []
### Iterate over objects
if AlignedWhite=='yes':
cmd.iterate(tObj1 + " and n. CA and not " + aln, "stored.alnAres.append(resi)")
cmd.iterate(tObj2 + " and n. CA and not " + aln, "stored.alnBres.append(resi)")
else:
cmd.iterate(tObj1 + " and n. CA", "stored.alnAres.append(resi)")
cmd.iterate(tObj2 + " and n. CA", "stored.alnBres.append(resi)")
### Change the B-factors for EACH object
displacementUpdateB(tObj1,stored.alnAres,tObj2,stored.alnBres)
### Store the NEW B-factors
stored.alnAnb, stored.alnBnb = [], []
### Iterate over objects and get b
if AlignedWhite=='yes':
### Iterate over objects which is not aligned
cmd.iterate(tObj1 + " and n. CA and not " + aln, "stored.alnAnb.append(b)" )
cmd.iterate(tObj2 + " and n. CA and not " + aln, "stored.alnBnb.append(b)" )
else:
### Or Iterate over all objects with CA
cmd.iterate(tObj1 + " and n. CA", "stored.alnAnb.append(b)" )
cmd.iterate(tObj2 + " and n. CA", "stored.alnBnb.append(b)" )
### Get rid of all intermediate objects and clean up
cmd.delete(tObj1)
cmd.delete(tObj2)
cmd.delete(aln)
### Assign the just stored NEW B-factors to the original objects
for x in range(len(stored.alnAres)):
cmd.alter(objSel1 + " and n. CA and i. " + str(stored.alnAres[x]), "b = " + str(stored.alnAnb[x]))
for x in range(len(stored.alnBres)):
cmd.alter(objSel2 + " and n. CA and i. " + str(stored.alnBres[x]), "b = " + str(stored.alnBnb[x]))
cmd.rebuild(); cmd.refresh(); cmd.sort(objSel1); cmd.sort(objSel2)
### Provide some useful information
stored.allRMSDval = []
stored.allRMSDval = stored.alnAnb + stored.alnBnb
print "\nColorByDisplacementCA completed successfully."
print "The MAXIMUM Displacement is: "+str(max(stored.allRMSDval)) +" residue "+str(stored.alnAres[int(stored.allRMSDval.index(max(stored.allRMSDval)))])
if strTrue(doColor):
### Showcase what we did
#cmd.orient()
#cmd.hide("all")
cmd.show("cartoon", objSel1 + " or " + objSel2)
### Select the residues not used for alignment; they still have their B-factors as "-0.2"
cmd.select("notUsedForAln", "b = -0.2")
### White-wash the residues not used for alignment
cmd.color("white", "notUsedForAln")
### Select the residues not in both pdb files; they have their B-factors as "-0. 01"
cmd.select("ResNotInBothPDB", "b = -0.01")
### White-wash the residues not used for alignment
cmd.color("black", "ResNotInBothPDB")
### Color the residues used for alignment according to their B-factors (Displacment values)
# cmd.spectrum("b", 'rainbow', "((" + objSel1 + " and n. CA) or (n. CA and " + objSel2 +" )) and not notUsedForAln+ResNotInBothPDB")
cmd.spectrum("b", 'rainbow', "((" + objSel1 + " and n. CA) or (n. CA and " + objSel2 +" )) and not (notUsedForAln or ResNotInBothPDB)")
### Delete the selection of atoms not used for alignment
### If you would like to keep this selection intact,
### just comment "cmd.delete" line and
### uncomment the "cmd.disable" line abowe.
cmd.disable("notUsedForAln")
cmd.delete("notUsedForAln")
cmd.disable("ResNotInBothPDB")
cmd.delete("ResNotInBothPDB")
print "\nObjects are now colored by C-alpha displacement deviation."
print "Blue is minimum and red is maximum..."
print "White is those residues used in the alignment algorithm. Can be turned off in top of algorithm."
print "Black is residues that does not exist in both files..."
cmd.extend("ColorByDisplacementCA", ColorByDisplacementCA)
def displacementUpdateBAll(objA, alnAri, objB, alnBri):
print "This will take a while to go through the for loops. Give me around 3-5 minutes..."
### If residue is unassigned in one of the pdb files, we reset its value
for x in range(len(alnAri)):
s1 = objA + " and resi " + alnAri[x][0] + " and name " + str(alnAri[x][1])
cmd.alter( s1, "b = " + str(-0.01))
for x in range(len(alnBri)):
s2 = objB + " and resi " + alnBri[x][0] + " and name " + alnBri[x][1]
cmd.alter( s2, "b = " + str(-0.01))
cmd.sort(objA); cmd.sort(objB)
for x in range(len(alnAri)):
s1 = objA + " and resi " + alnAri[x][0] + " and name " + alnAri[x][1]
s2 = objB + " and resi " + alnAri[x][0] + " and name " + alnAri[x][1]
### Names starting with __ (underscores) are normally hidden by PyMOL
tempObject = "__tempObject"
Displacement = cmd.distance(tempObject, s1, s2)
cmd.alter( s1, "b = " + str(Displacement))
cmd.alter( s2, "b = " + str(Displacement))
cmd.delete(tempObject)
cmd.sort(objA); cmd.sort(objB)
def ColorByDisplacementAll(objSel1, objSel2, super1='all', super2='all', doColor="True", doAlign="True", AlignedWhite='yes'):
### First create backup copies; names starting with __ (underscores) are normally hidden by PyMOL
tObj1, tObj2, aln = "__tempObj1", "__tempObj2", "__aln"
if strTrue(doAlign):
### Create temp objects
cmd.create( tObj1, objSel1 )
cmd.create( tObj2, objSel2 )
### Align and make create an object aln which indicates which atoms were paired between the two structures
### Super is must faster than align http://www.pymolwiki.org/index.php/Super
cmd.super(tObj1 + ' and ' + str(super1), tObj2 + ' and ' + str(super2), object=aln)
### Modify the original matrix of object1 from the alignment
cmd.matrix_copy(tObj1, objSel1)
else:
### Create temp objects
cmd.create( tObj1, objSel1 )
cmd.create( tObj2, objSel2 )
### Align and make create an object aln which indicates which atoms were paired between the two structures
### Super is must faster than align http://www.pymolwiki.org/index.php/Super
cmd.super(tObj1 + ' and ' + str(super1), tObj2 + ' and ' + str(super2), object=aln)
### Modify the B-factor columns of the original objects,
### in order to identify the residues NOT used for alignment, later on
cmd.alter( objSel1 + " or " + objSel2, "b=-0.2")
cmd.alter( tObj1 + " or " + tObj2, "chain='A'")
cmd.alter( tObj1 + " or " + tObj2, "segi='A'")
### Update pymol internal representations; one of these should do the trick
cmd.refresh(); cmd.rebuild(); cmd.sort(tObj1); cmd.sort(tObj2)
### Create lists for storage
stored.alnAres, stored.alnBres = [], []
### Iterate over objects and get resi
if AlignedWhite=='yes':
cmd.iterate(tObj1 + " and not " + aln, "stored.alnAres.append((resi, name))")
cmd.iterate(tObj2 + " and not " + aln, "stored.alnBres.append((resi, name))")
else:
cmd.iterate(tObj1, "stored.alnAres.append((resi, name))")
cmd.iterate(tObj2, "stored.alnBres.append((resi, name))")
### Change the B-factors for EACH object
displacementUpdateBAll(tObj1,stored.alnAres,tObj2,stored.alnBres)
### Store the NEW B-factors
stored.alnAnb, stored.alnBnb = [], []
### Iterate over objects and get b
if AlignedWhite=='yes':
### Iterate over objects which is not aligned
cmd.iterate(tObj1 + " and not " + aln, "stored.alnAnb.append(b)" )
cmd.iterate(tObj2 + " and not " + aln, "stored.alnBnb.append(b)" )
else:
### Or Iterate over all objects with CA
cmd.iterate(tObj1, "stored.alnAnb.append(b)" )
cmd.iterate(tObj2, "stored.alnBnb.append(b)" )
### Get rid of all intermediate objects and clean up
cmd.delete(tObj1)
cmd.delete(tObj2)
cmd.delete(aln)
### Assign the just stored NEW B-factors to the original objects
print "Sooon ready. 1 more minute"
for x in range(len(stored.alnAres)):
cmd.alter(objSel1 + " and resi " + str(stored.alnAres[x][0]) + " and name " + str(stored.alnAres[x][1]), "b = " + str(stored.alnAnb[x]))
for x in range(len(stored.alnBres)):
cmd.alter(objSel2 + " and resi " + str(stored.alnBres[x][0]) + " and name " + str(stored.alnBres[x][1]), "b = " + str(stored.alnBnb[x]))
cmd.rebuild(); cmd.refresh(); cmd.sort(objSel1); cmd.sort(objSel2)
### Provide some useful information
stored.allRMSDval = []
stored.allRMSDval = stored.alnAnb + stored.alnBnb
print "\nColorByDisplacementAll completed successfully."
print "The MAXIMUM Displacement is: "+str(max(stored.allRMSDval)) +" residue "+str(stored.alnAres[int(stored.allRMSDval.index(max(stored.allRMSDval)))])
if strTrue(doColor):
### Showcase what we did
#cmd.orient()
#cmd.hide("all")
cmd.show("sticks", objSel1 + " or " + objSel2)
### Select the residues not used for alignment; they still have their B-factors as "-0.2"
cmd.select("notUsedForAln", "b = -0.2")
### White-wash the residues not used for alignment
cmd.color("white", "notUsedForAln")
### Select the residues not in both pdb files; they have their B-factors as "-0.01"
cmd.select("ResNotInBothPDB", "b = -0.01")
### White-wash the residues not used for alignment
cmd.color("black", "ResNotInBothPDB")
### Color the residues used for alignment according to their B-factors (Displacement values)
# cmd.spectrum("b", 'rainbow', "((" + objSel1 + ") or (" + objSel2 +" )) and not notUsedForAln+ResNotInBothPDB")
cmd.spectrum("b", 'rainbow', "((" + objSel1 + ") or (" + objSel2 +" )) and not (notUsedForAln or ResNotInBothPDB)")
### Delete the selection of atoms not used for alignment
### If you would like to keep this selection intact,
### just comment "cmd.delete" line and
### uncomment the "cmd.disable" line abowe.
cmd.disable("notUsedForAln")
cmd.delete("notUsedForAln")
cmd.disable("ResNotInBothPDB")
cmd.delete("ResNotInBothPDB")
print "\nObjects are now colored by C-alpha displacement deviation."
print "Blue is minimum and red is maximum..."
print "White is those residues used in the alignment algorithm. Can be turned off in top of algorithm."
print "Black is residues that does not exist in both files..."
cmd.extend("ColorByDisplacementAll", ColorByDisplacementAll)