ColorByRMSD: Difference between revisions
Jump to navigation
Jump to search
m (→Code) |
(rewrite of script and moved script to github) |
||
| (12 intermediate revisions by one other user not shown) | |||
| Line 1: | Line 1: | ||
{{Infobox script-repo | |||
|type = Python Module | |||
|filename = colorbyrmsd.py | |||
|author = [[User:Shiven|Shivender Shandilya]], [[User:Inchoate|Jason Vertrees]], [[User:Speleo3|Thomas Holder]] | |||
|license = BSD-2-Clause | |||
}} | |||
== Introduction == | == Introduction == | ||
This script allows you to color two structures by Root Mean Square Deviation (RMSD). | |||
The distances between aligned C-alpha atom pairs are stored as B-factors of these residues, which are colored by a color spectrum, with blue specifying the minimum pairwise RMSD and red indicating the maximum. | |||
Unaligned residues are colored gray. | |||
== Usage == | |||
colorbyrmsd mobile, target [, doAlign [, doPretty [, guide [, method ]]]] | |||
== Arguments == | |||
= | * '''mobile''' = string: atom selection for mobile atoms | ||
* '''target''' = string: atom selection for target atoms | |||
* '''doAlign''' = 0 or 1: Superpose selections before calculating distances {default: 1} | |||
* '''doPretty''' = 0 or 1: Show nice representation and colors {default: 1} | |||
* '''guide''' = 0 or 1: Only use C-alpha atoms {default: 1} | |||
* '''method''' = align or super: Method to match atoms {default: super} | |||
== Examples == | |||
<source lang="python"> | <source lang="python"> | ||
# example #1 | # example #1 | ||
colorbyrmsd 1cbs, 1hmt, doAlign=1, doPretty=1 | |||
# example #2 | # example #2 | ||
colorbyrmsd 1eaz, 1fao, doAlign=1, doPretty=1 | |||
</source> | </source> | ||
<gallery heights="300px" widths="300px"> | <gallery heights="300px" widths="300px"> | ||
Image:ColorByRMSD_1cbs_1hmt.png|1cbs and 1hmt aligned and colored by RMSD. Dark blue is good alignment, higher deviations are in orange/yellow/red. | Image:ColorByRMSD_1cbs_1hmt.png|1cbs and 1hmt aligned and colored by RMSD. Dark blue is good alignment, higher deviations are in orange/yellow/red. Residues not used for alignment are colored white. | ||
Image:ColorByRMSD_1eaz_1fao.png|1eaz and 1fao aligned and colored by RMSD. Dark blue is good alignment, higher deviations are in orange/yellow/red. | Image:ColorByRMSD_1eaz_1fao.png|1eaz and 1fao aligned and colored by RMSD. Dark blue is good alignment, higher deviations are in orange/yellow/red. Residues not used for alignment are colored white. | ||
</gallery> | </gallery> | ||
[[Category:Script_Library]] | [[Category:Script_Library]] | ||
[[Category:Structural_Biology_Scripts]] | [[Category:Structural_Biology_Scripts]] | ||
[[Category:Pymol-script-repo]] | |||
Latest revision as of 08:59, 16 March 2012
| Type | Python Module |
|---|---|
| Download | colorbyrmsd.py |
| Author(s) | Shivender Shandilya, Jason Vertrees, Thomas Holder |
| License | BSD-2-Clause |
| This code has been put under version control in the project Pymol-script-repo | |
Introduction
This script allows you to color two structures by Root Mean Square Deviation (RMSD). The distances between aligned C-alpha atom pairs are stored as B-factors of these residues, which are colored by a color spectrum, with blue specifying the minimum pairwise RMSD and red indicating the maximum. Unaligned residues are colored gray.
Usage
colorbyrmsd mobile, target [, doAlign [, doPretty [, guide [, method ]]]]
Arguments
- mobile = string: atom selection for mobile atoms
- target = string: atom selection for target atoms
- doAlign = 0 or 1: Superpose selections before calculating distances {default: 1}
- doPretty = 0 or 1: Show nice representation and colors {default: 1}
- guide = 0 or 1: Only use C-alpha atoms {default: 1}
- method = align or super: Method to match atoms {default: super}
Examples
# example #1
colorbyrmsd 1cbs, 1hmt, doAlign=1, doPretty=1
# example #2
colorbyrmsd 1eaz, 1fao, doAlign=1, doPretty=1
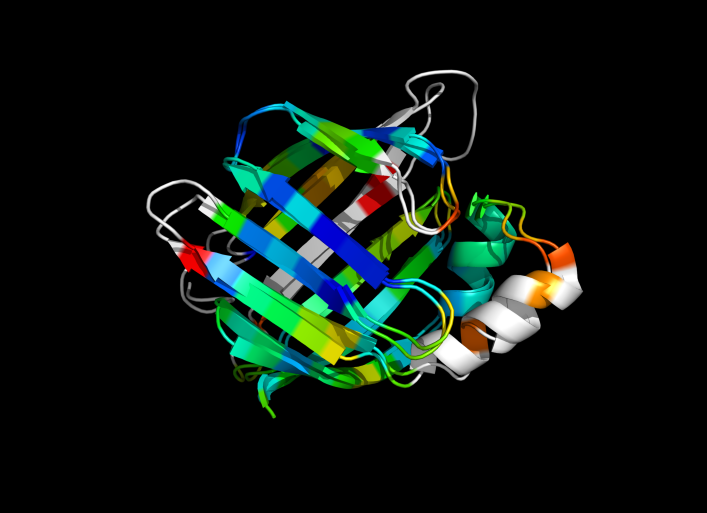
1cbs and 1hmt aligned and colored by RMSD. Dark blue is good alignment, higher deviations are in orange/yellow/red. Residues not used for alignment are colored white.
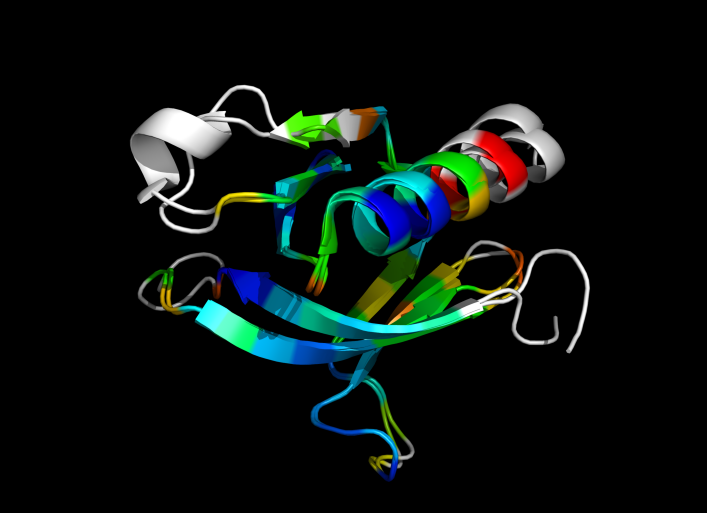
1eaz and 1fao aligned and colored by RMSD. Dark blue is good alignment, higher deviations are in orange/yellow/red. Residues not used for alignment are colored white.

