Difference between revisions of "Split states"
Jump to navigation
Jump to search
m (Small typo) |
|||
| Line 1: | Line 1: | ||
| − | |||
'''Split_States''' splits and orients multiple models and multimers from the biological unit file into a set of single-state molecular objects. | '''Split_States''' splits and orients multiple models and multimers from the biological unit file into a set of single-state molecular objects. | ||
| Line 7: | Line 6: | ||
</source> | </source> | ||
| − | This splits the '''object''' from '''first''' to '''last''' out to the array of objects prefixed by '''prefix'''. The '''prefix''' option is very handy | + | This splits the '''object''' from '''first''' to '''last''' out to the array of objects prefixed by '''prefix'''. The '''prefix''' option is very handy if all your states--or a subset of the states--have the same name. |
==Using== | ==Using== | ||
Revision as of 04:21, 7 January 2010
Split_States splits and orients multiple models and multimers from the biological unit file into a set of single-state molecular objects.
Syntax
split_states object [, first [, last [, prefix ]]]
This splits the object from first to last out to the array of objects prefixed by prefix. The prefix option is very handy if all your states--or a subset of the states--have the same name.
Using
To use split_states simply Load your molecule
# example usage
load fileName.pdb1, name
split_states name
delete name
# split all the states to objects starting with conf
fetch 1nmr
split_states 1nmr, prefix=conf
Example
1VLS: A dimer.
load 1vls.pdb1, 1vls
split_states 1vls
dele 1vls

1VLS as a monomer. This is the state of 1VLS when I load the molecule (and select cartoon representation).
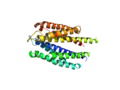
1VLS as a dimer using the split_states command. Notice PyMOL automatically loads and orients the new molecules.

