Difference between revisions of "Propka"
| (96 intermediate revisions by the same user not shown) | |||
| Line 1: | Line 1: | ||
| + | {{Infobox script-repo | ||
| + | |type = script | ||
| + | |filename = propka.py | ||
| + | |author = [[User:Tlinnet|Troels E. Linnet]] | ||
| + | |license = BSD | ||
| + | }} | ||
| + | |||
== Acknowledgement == | == Acknowledgement == | ||
| − | + | propka.py contact and relies on the result from the [http://propka.ki.ku.dk propka] server | |
| − | + | The PROPKA method is developed by the [http://propka.ki.ku.dk/~luca/wiki/index.php/JansPage Jensen Research Group] , Department of Chemistry, University of Copenhagen. <br /> | |
== Introduction == | == Introduction == | ||
| Line 8: | Line 15: | ||
It also automatically writes a pymol command file and let pymol execute it. This command file make pka atoms, rename them, label them and color them according to the pka value. | It also automatically writes a pymol command file and let pymol execute it. This command file make pka atoms, rename them, label them and color them according to the pka value. | ||
| − | If is | + | If you put the mechanize folder and the propka.py script somewhere in your pymol search path, then getting the pka values is made super easy. By the way, did you know, that you don't have to prepare the .pdb file by adding/removing hydrogens? The [http://propka.ki.ku.dk/ propka] server uses its own internal hydrogen placement algorithm. |
| − | + | ||
| + | <syntaxhighlight lang="python"> | ||
| + | import propka | ||
| + | fetch 4ins, async=0 | ||
| + | propka | ||
| + | </syntaxhighlight> | ||
| + | |||
| + | If there is no web connection, it is possible to process a result file from a previous run or from a downloaded propka webpage result. This can be a handsome feature in a teaching/seminar situation, since it speeds up the pymol result or that an available web connection can be doubtful. Just point to the .pka file: Remember the dot "." which means "current directory". | ||
| + | <syntaxhighlight lang="python"> | ||
| + | import propka | ||
| + | load 4ins.pdb | ||
| + | propka pkafile=./Results_propka/4ins"LOGTIME".pka, resi=18.25-30, resn=cys | ||
| + | </syntaxhighlight> | ||
The last possibility, is just to ask for the pka values of a recognized PDB id. This is done with the "getpropka" function. | The last possibility, is just to ask for the pka values of a recognized PDB id. This is done with the "getpropka" function. | ||
| + | <syntaxhighlight lang="python"> | ||
| + | import propka | ||
| + | getpropka source=ID, PDBID=4ake, logtime=_, showresult=yes | ||
| + | </syntaxhighlight> | ||
== Dependency of python module: mechanize == | == Dependency of python module: mechanize == | ||
| − | The script needs mechanize to run. | + | The script needs mechanize to run. The module is included in the project, [http://www.pymolwiki.org/index.php/Git_intro Pymol-script-repo]. |
* On windows, it is not easy to make additional modules available for pymol. So put in into your working folder. | * On windows, it is not easy to make additional modules available for pymol. So put in into your working folder. | ||
* The easy manual way: | * The easy manual way: | ||
| Line 24: | Line 47: | ||
=== Examples === | === Examples === | ||
| − | < | + | Read about the proteins here:<br> |
| − | + | http://www.proteopedia.org/wiki/index.php/4ins <br> | |
| − | + | http://www.proteopedia.org/wiki/index.php/1hp1 <br> | |
| − | + | ||
| + | <syntaxhighlight lang="python"> | ||
| + | import propka | ||
| − | |||
fetch 4ins, async=0 | fetch 4ins, async=0 | ||
| − | propka | + | propka OR |
| + | propka 4ins OR | ||
| + | propka 4ins, resi=19.20, resn=ASP.TYR, logtime=_, verbose=yes | ||
| + | import propka | ||
fetch 1hp1, async=0 | fetch 1hp1, async=0 | ||
| − | propka molecule=1hp1, chain=A, resi=308.513, resn=CYS, logtime=_ | + | propka molecule=1hp1, chain=A, resi=305-308.513, resn=CYS, logtime=_ |
| − | getpropka source=ID, PDBID=4ins, logtime=_, server_wait=3.0, verbose=yes, showresult= | + | import propka |
| − | </ | + | getpropka source=ID, PDBID=4ins, logtime=_, server_wait=3.0, verbose=yes, showresult=yes |
| + | </syntaxhighlight> | ||
<gallery heights="240px" widths="340px"> | <gallery heights="240px" widths="340px"> | ||
| − | Image: | + | Image:Propka1HP1.png|propka used on 1HP1. |
| − | Image: | + | Image:Propka1HP1-ATP.png|propka used on 1HP1, zoom on ATP ligand. |
| − | Image: | + | Image:Propka1HP1-ZN.png|propka used on 1HP1, zoom on ZN ligand metal center. |
</gallery> | </gallery> | ||
| − | |||
<gallery heights="240px" widths="340px"> | <gallery heights="240px" widths="340px"> | ||
| − | Image: | + | Image:Propka4INS.png|propka used on 4INS. |
| − | Image: | + | Image:Pymolpropkamenu.png|The easy menu does it easy to click on/off ligands and bonds |
| − | Image: | + | Image:Resultslog.png|An appending logfile ./Results_propka/_Results.log saves the input commands and the result for the residues specified with resi= and resn= |
</gallery> | </gallery> | ||
| + | |||
pka atoms are created and renamed for their pka value. That makes it easy to "click" the atom in pymol and instantly see the pka value. | pka atoms are created and renamed for their pka value. That makes it easy to "click" the atom in pymol and instantly see the pka value. | ||
| Line 58: | Line 86: | ||
If one wants to see the specified result, the logfile ./Results_propka/_Results.log saves the link to the propka server. Here one can see in an interactive Jmol appp, the interactions to the pka residues. | If one wants to see the specified result, the logfile ./Results_propka/_Results.log saves the link to the propka server. Here one can see in an interactive Jmol appp, the interactions to the pka residues. | ||
| − | == | + | == Use of script == |
| − | < | + | <syntaxhighlight lang="python"> |
| − | + | cd /home/tlinnet/test | |
| − | |||
| − | |||
| − | + | import propka | |
| − | |||
| − | |||
| − | |||
| − | ### | + | ### The fastest method is just to write propka. Then the last pymol molecule is assumed and send to server. verbose=yes makes the script gossip mode. |
fetch 4ins, async=0 | fetch 4ins, async=0 | ||
| − | propka | + | propka |
| − | ### | + | ### Larger protein |
| − | + | fetch 1hp1, async=0 | |
| − | + | propka logtime=_, resi=5-10.20-30, resn=CYS.ATP.TRP, verbose=yes | |
| − | |||
| − | |||
| − | |||
| − | ### | + | ### Fetch 4ins from web. async make sure, we dont execute script before molecule is loaded. The resi and resn prints the interesting results right to command line. |
| − | + | fetch 4ins, async=0 | |
| − | + | propka chain=*, resi=5-10.20-30, resn=ASP.CYS, logtime=_ | |
| − | ### | + | ### If there is no web connection, one can process a local .pka file. Either from a previous run or from a downloaded propka webpage result. |
| − | + | ### Then run and point to .pka file with: pkafile=./Results_propka/pkafile.pka Remember the dot "." in the start, to make it start in the current directory. | |
| − | + | load 4ins.pdb | |
| − | + | propka pkafile=./Results_propka/4ins_.pka, resi=18.25-30, resn=cys, | |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | # | ||
| − | # | ||
| − | # | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | = | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | ### Some more examples. This molecule has 550 residues, so takes a longer time. We select to run the last molecule, by writing: molecule=1hp1 | |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | # | ||
| − | # | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | run | ||
| − | |||
| − | |||
| − | |||
| − | |||
fetch 4ins, async=0 | fetch 4ins, async=0 | ||
| − | propka molecule= | + | fetch 1hp1, async=0 |
| + | propka molecule=1hp1, chain=A, resi=300-308.513, resn=CYS.ATP.TRP, logtime=_, verbose=no, showresult=no | ||
| + | propka molecule=1hp1, pkafile=./Results_propka/1hp1_.pka, verbose=yes | ||
| + | </syntaxhighlight> | ||
| − | ### | + | === Input paramaters === |
| − | ### | + | <syntaxhighlight lang="text"> |
| − | # | + | ############################################Input parameters: propka############################################ |
| − | # | + | ############# The order of input and changable things: |
| − | # | + | propka(molecule="NIL",chain="*",resi="0",resn="NIL",method="upload",logtime=time.strftime("%m%d",time.localtime()),server_wait=3.0,version="v3.1",verbose="no",showresult="no",pkafile="NIL") |
| − | # | ||
| − | |||
| − | ### | ||
| − | # | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
# method : method=upload is default. This sends .pdb file and request result from propka server. | # method : method=upload is default. This sends .pdb file and request result from propka server. | ||
## method=file will only process a manual .pka file, and write a pymol command file. No use of mechanize. | ## method=file will only process a manual .pka file, and write a pymol command file. No use of mechanize. | ||
| + | ## If one points to an local .pka file, then method is auto-changed to method=file. This is handsome in off-line environment, ex. teaching or seminar. | ||
| + | # pkafile: Write the path to .pka file. Ex: pkafile=./Results_propka/4ins_.pka | ||
# molecule : name of the molecule. Ending of file is assumed to be .pdb | # molecule : name of the molecule. Ending of file is assumed to be .pdb | ||
# chain : which chains are saved to file, before molecule file is send to server. Separate with "." Ex: chain=A.b | # chain : which chains are saved to file, before molecule file is send to server. Separate with "." Ex: chain=A.b | ||
| − | # resi : Select by residue number, which residues should be printed to screen and saved to the log file: | + | # resi : Select by residue number, which residues should be printed to screen and saved to the log file: /Results_propka/_Results.log. |
## Separate with "." or make ranges with "-". Ex: resi=35.40-50 | ## Separate with "." or make ranges with "-". Ex: resi=35.40-50 | ||
| − | # resn : Select by residue name, which residues should be printed to screen and saved to the log file: | + | # resn : Select by residue name, which residues should be printed to screen and saved to the log file: /Results_propka/_Results.log. |
## Separate with "." Ex: resn=cys.tyr | ## Separate with "." Ex: resn=cys.tyr | ||
# logtime : Each execution give a set of files with the job id=logtime. If logtime is not provided, the current time is used. | # logtime : Each execution give a set of files with the job id=logtime. If logtime is not provided, the current time is used. | ||
| Line 212: | Line 135: | ||
# verbose : Verbose is switch, to turn on messages for the mechanize section. This is handsome to see how mechanize works, and for error searching. | # verbose : Verbose is switch, to turn on messages for the mechanize section. This is handsome to see how mechanize works, and for error searching. | ||
# showresult : Switch, to turn on all results in pymol command window. Ex: showresult=yes | # showresult : Switch, to turn on all results in pymol command window. Ex: showresult=yes | ||
| − | |||
# server_wait=10.0 is default. This defines how long time between asking the server for a result. Set no lower than 3 seconds. | # server_wait=10.0 is default. This defines how long time between asking the server for a result. Set no lower than 3 seconds. | ||
# version=v3.1 is default. This is what version of propka which would be used. | # version=v3.1 is default. This is what version of propka which would be used. | ||
## Possible: 'v3.1','v3.0','v2.0'. If a newer version is available than the current v3.1, a error message is raised to make user update the script. | ## Possible: 'v3.1','v3.0','v2.0'. If a newer version is available than the current v3.1, a error message is raised to make user update the script. | ||
| − | # source : source=upload is default and is set at the propka webpage | + | ############################################Input parameters: getpropka############################################ |
| − | # | + | ############# The order of input and changable things: |
| + | getpropka(PDB="NIL",chain="*",resi="0",resn="NIL",source="upload",PDBID="",logtime=time.strftime("%Y%m%d%H%M%S",time.localtime()),server_wait=3.0,version="v3.1",verbose="no",showresult="no") | ||
| + | # PDB: points the path to a .pdb file. This is auto-set from propka function. | ||
| + | # source : source=upload is default and is set at the propka webpage. | ||
| + | # source=ID, PDBID=4ake , one can print to the command line, the pka value for any official pdb ID. No files are displayed in pymol. | ||
| + | # PDBID: is used as the 4 number/letter pdb code, when invoking source=ID. | ||
| + | </syntaxhighlight> | ||
| + | |||
| + | == Examples == | ||
| + | === Example 1 - Mutagenesis analysis - part 1 === | ||
| + | This script was developed with the intention of making analysis of possible mutants easier. For example, the reactivity of Cysteines in FRET maleimide labelling is determined by the fraction of the Cysteine residue which is negatively charged (C-). This fraction is related to its pKa value and the pH of the buffer: f(C-)=1/(10(pK-pH)+1). So, one would be interested in having the lowest possible pKa value as possible. Ideally lower than the pH of the buffer. To analyse where to make the best mutant in your protein, you could do the following for several residues. We do the mutagenesis in the command line, since we then could loop over the residues in the protein.<br> | ||
| + | So, in a loop with defined residues, this could look like the following code. Note, now we are quite happy for the result log file, since it collects the pka for the mutants. | ||
| + | |||
| + | {{Template:PymolScriptRepoDownload|examples/propka_1.pml}} | ||
| + | <include src="https://raw.github.com/Pymol-Scripts/Pymol-script-repo/master/examples/propka_1.pml" highlight="python" /> | ||
| + | |||
| + | === Example 2 - Mutagenesis analysis - part 2 === | ||
| + | To only loop over surface residues, you might want to find these with the script [[FindSurfaceResidues]]. | ||
| − | + | {{Template:PymolScriptRepoDownload|examples/propka_2.pml}} | |
| + | <include src="https://raw.github.com/Pymol-Scripts/Pymol-script-repo/master/examples/propka_2.pml" highlight="python" /> | ||
| − | + | A little warning though. You need to be carefull about the rotamer you're choosing. It can happen that the first rotamer ends up being in physically non-reasonable contact distance to other residues, so atoms become overlayed. Also, the mutagenesis wizard can have the funny habit of sometimes not adding hydrogens to terminal -C or -N after mutating a residue. | |
| − | + | === Example 3 - Scan a range of proteins === | |
| − | + | Could be done with this script | |
| − | |||
| − | + | {{Template:PymolScriptRepoDownload|examples/propka_3.pml}} | |
| − | + | <include src="https://raw.github.com/Pymol-Scripts/Pymol-script-repo/master/examples/propka_3.pml" highlight="python" /> | |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | == ScriptVersion == | |
| − | + | Current_Version=20111202 | |
| − | |||
| − | + | == Changelog == | |
| − | + | * 20111202 | |
| − | + | *# This code has been put under version control. In the project, [http://www.pymolwiki.org/index.php/Git_intro Pymol-script-repo]. | |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | * 20110823 | |
| − | + | *# Fixed some issues with selection algebra of Ligands. | |
| − | + | *# Now colors Ligands automatically to purple scheme. | |
| − | |||
| − | + | * 20110822 | |
| − | + | *# Made the naming scheme consistent, so one can work with multiple proteins, and the grouping still works. | |
| − | + | *# Bonds to N-terminal and C-terminal did not show up. Fixed. | |
| − | + | *# If one just write "propka", the "last" molecule in the pymol object list is now assumed, instead of the first. This makes mutagenesis analysis easier. | |
| − | + | *# The pka difference from assumed standard values are now also displayed. Standard values are set to: pkadictio = {'ASP':3.9, 'GLU':4.3, 'ARG':12.0, 'LYS':10.5, 'HIS':6.0, 'CYS':8.3, 'TYR':10.1} | |
| − | + | *# The menu size is made bigger, so it can fit the long names for the bonding partners. There's also a slider that you can drag using the mouse | |
| − | + | *#:--it's the tiny tab between the command line and the movie controls. Drag that left or right. | |
| − | + | * 20110821 | |
| − | + | *# The resn function was made wrong, only taking the last item of list. Fixed. | |
| − | + | *# resn= can now also be print to screen/log for ligands like ATP. Ex: resn=CYS.ATP.TRP | |
| − | + | *# Ligands are now also written to the "stripped-file". "./Results_propka/PDB.stripped" | |
| − | + | *# Bonds are now also made for Ligands. | |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | * 20110820 | |
| − | + | *# If one points to a result .pka file, then "method" is automatically set to "method=file". | |
| − | + | *# Added the ability for pka values for ligands | |
| − | + | *# Bonds are now generated for the pka atoms. | |
| − | + | *# The color scheme is changed from "rainbow" to "red_white_blue". This is easier to interpret. | |
| + | ** CC: COULOMBIC INTERACTION. Color is red. | ||
| + | ** SH: SIDECHAIN HYDROGEN BOND. Color is brightorange. | ||
| + | ** BH: BACKBONE HYDROGEN BOND. Color is lightorange. | ||
| − | + | * 20110817 | |
| − | + | *# If just invoking with "propka", it will select the first molecule. And now it is possible also to write. "propka all". | |
| − | + | *# Removed the "raise UserWarning" if the script is oudated. Only a warning message is printed. | |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | * 20110816 | |
| − | + | *# Made the execution of the pymol command script silent, by only using cmd.API. This will raise a Warning, which can be ignored. | |
| − | + | *# Built-in a Script version control, to inform the user if the propka script is updated on this page | |
| − | + | *# The alternate attribute for the labeling atoms are reset. It was found pymol objected altering names for atoms which had alternate positions/values. | |
| − | + | *# Reorganized the input order, which means that: molecule=all is default. | |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | === Known bugs === | |
| − | + | * Bonds can be multiplied from amino acids to Ligands like ZN or ATP. Assume the shortest bond to be correct. | |
| − | + | *# ZN: This is caused, since ZN has no ID number and when there are several in the same chain. This can be reproduced for 1HP1, bond: A41ZNCC. Here it shows to bonds, where only the shortes is correct. No fix possible. | |
| + | *# ATP: This is caused, since propka sometimes eats the ending of the atom name. In the .pdb file is written O3', which propka represents like O3. Therefore a wildcard "*" is generel inserted, which can cause multiple bonds to the ATP molecule. This can be reproduced for 1HP1, bond: A504ATPSH. Here multiple bonds are made to all the O3*,O2* atoms. No fix possible. | ||
| + | * The propka server sometimes use another naming scheme for Ligands. These Ligands will not be pka labelled or shown in pymol. The results will still downloaded to "/.Results_propka/file.pka" | ||
| + | *# This can be reproduced with 1BJ6. Here the propka webpage predicts the pka values for the ligands: AN7,GN1,CN3,CO2,GO2, but the .pdb file names these ligands: DA,DA,DC,DG. | ||
| + | * Alternative configurations of a Ligand is at the moment a problem, and will not be shown. For example 1HXB and the ligand ROC. | ||
| + | *: The script extraxt AROC and BROC from pymol, which does match with ROC in the .pka file. Try save the protein with only one configuration of the Ligand. | ||
| + | ** >In pymol: | ||
| + | **: fetch 1hxb, async=0 | ||
| + | **: create 1hxbA, 1hxb and not alt B | ||
| + | **: save 1hxbA.pdb, 1hxbA | ||
| + | ** >Quit pymol | ||
| + | ** >Manually replace with text editor in .pdb file "AROC" with " ROC" Remember the space " "! | ||
| + | ** >Then in pymol | ||
| + | **: load 1hxbA.pdb | ||
| + | **: import propka | ||
| + | **: propka verbose=yes | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
[[Category:Script_Library]] | [[Category:Script_Library]] | ||
[[Category:Structural_Biology_Scripts]] | [[Category:Structural_Biology_Scripts]] | ||
[[Category:Biochemical_Scripts]] | [[Category:Biochemical_Scripts]] | ||
| + | [[Category:Pymol-script-repo]] | ||
Latest revision as of 11:06, 15 January 2012
| Type | Python Script |
|---|---|
| Download | propka.py |
| Author(s) | Troels E. Linnet |
| License | BSD |
| This code has been put under version control in the project Pymol-script-repo | |
Acknowledgement
propka.py contact and relies on the result from the propka server
The PROPKA method is developed by the Jensen Research Group , Department of Chemistry, University of Copenhagen.
Introduction
This script can fetch the pka values for a protein from the propka server. The "propka" function downloads the results and processes them.
It also automatically writes a pymol command file and let pymol execute it. This command file make pka atoms, rename them, label them and color them according to the pka value.
If you put the mechanize folder and the propka.py script somewhere in your pymol search path, then getting the pka values is made super easy. By the way, did you know, that you don't have to prepare the .pdb file by adding/removing hydrogens? The propka server uses its own internal hydrogen placement algorithm.
import propka
fetch 4ins, async=0
propka
If there is no web connection, it is possible to process a result file from a previous run or from a downloaded propka webpage result. This can be a handsome feature in a teaching/seminar situation, since it speeds up the pymol result or that an available web connection can be doubtful. Just point to the .pka file: Remember the dot "." which means "current directory".
import propka
load 4ins.pdb
propka pkafile=./Results_propka/4ins"LOGTIME".pka, resi=18.25-30, resn=cys
The last possibility, is just to ask for the pka values of a recognized PDB id. This is done with the "getpropka" function.
import propka
getpropka source=ID, PDBID=4ake, logtime=_, showresult=yes
Dependency of python module: mechanize
The script needs mechanize to run. The module is included in the project, Pymol-script-repo.
- On windows, it is not easy to make additional modules available for pymol. So put in into your working folder.
- The easy manual way:
- Go to: http://wwwsearch.sourceforge.net/mechanize/download.html
- Download mechanize-0.2.5.zip. http://pypi.python.org/packages/source/m/mechanize/mechanize-0.2.5.zip
- Extract to .\mechanize-0.2.5 then move the in-side folder "mechanize" to your folder with propka.py. The rest of .\mechanize-0.2.5 you don't need.
- You can also see other places where you could put the "mechanize" folder. Write this in pymol to see the paths where pymol is searching for "mechanize"
- import sys; print(sys.path)
Examples
Read about the proteins here:
http://www.proteopedia.org/wiki/index.php/4ins
http://www.proteopedia.org/wiki/index.php/1hp1
import propka
fetch 4ins, async=0
propka OR
propka 4ins OR
propka 4ins, resi=19.20, resn=ASP.TYR, logtime=_, verbose=yes
import propka
fetch 1hp1, async=0
propka molecule=1hp1, chain=A, resi=305-308.513, resn=CYS, logtime=_
import propka
getpropka source=ID, PDBID=4ins, logtime=_, server_wait=3.0, verbose=yes, showresult=yes
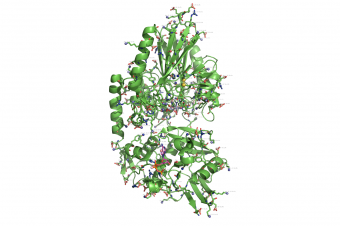
propka used on 1HP1.
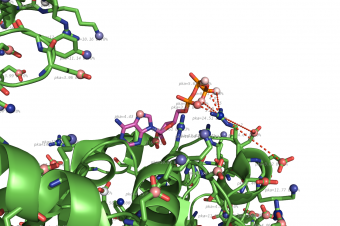
propka used on 1HP1, zoom on ATP ligand.
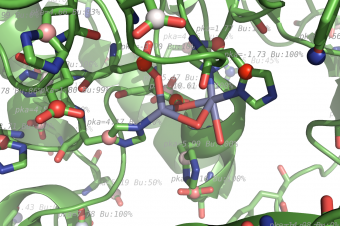
propka used on 1HP1, zoom on ZN ligand metal center.



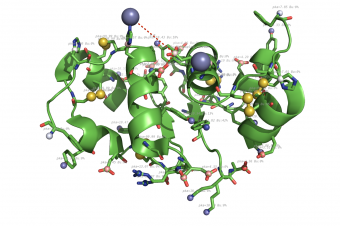
propka used on 4INS.
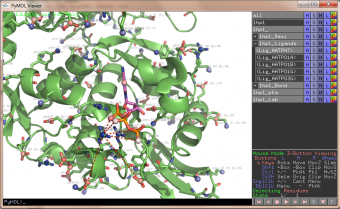
The easy menu does it easy to click on/off ligands and bonds
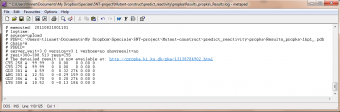
An appending logfile ./Results_propka/_Results.log saves the input commands and the result for the residues specified with resi= and resn=



pka atoms are created and renamed for their pka value. That makes it easy to "click" the atom in pymol and instantly see the pka value.
The atoms b value are also altered to the pka value, and the atoms are then spectrum colored from pka=0-14.
The pka value of 99.9 represent a di-sulphide bond, and is colored gold and the sphere size is set a little bigger.
If one wants to see the specified result, the logfile ./Results_propka/_Results.log saves the link to the propka server. Here one can see in an interactive Jmol appp, the interactions to the pka residues.
Use of script
cd /home/tlinnet/test
import propka
### The fastest method is just to write propka. Then the last pymol molecule is assumed and send to server. verbose=yes makes the script gossip mode.
fetch 4ins, async=0
propka
### Larger protein
fetch 1hp1, async=0
propka logtime=_, resi=5-10.20-30, resn=CYS.ATP.TRP, verbose=yes
### Fetch 4ins from web. async make sure, we dont execute script before molecule is loaded. The resi and resn prints the interesting results right to command line.
fetch 4ins, async=0
propka chain=*, resi=5-10.20-30, resn=ASP.CYS, logtime=_
### If there is no web connection, one can process a local .pka file. Either from a previous run or from a downloaded propka webpage result.
### Then run and point to .pka file with: pkafile=./Results_propka/pkafile.pka Remember the dot "." in the start, to make it start in the current directory.
load 4ins.pdb
propka pkafile=./Results_propka/4ins_.pka, resi=18.25-30, resn=cys,
### Some more examples. This molecule has 550 residues, so takes a longer time. We select to run the last molecule, by writing: molecule=1hp1
fetch 4ins, async=0
fetch 1hp1, async=0
propka molecule=1hp1, chain=A, resi=300-308.513, resn=CYS.ATP.TRP, logtime=_, verbose=no, showresult=no
propka molecule=1hp1, pkafile=./Results_propka/1hp1_.pka, verbose=yes
Input paramaters
############################################Input parameters: propka############################################
############# The order of input and changable things:
propka(molecule="NIL",chain="*",resi="0",resn="NIL",method="upload",logtime=time.strftime("%m%d",time.localtime()),server_wait=3.0,version="v3.1",verbose="no",showresult="no",pkafile="NIL")
# method : method=upload is default. This sends .pdb file and request result from propka server.
## method=file will only process a manual .pka file, and write a pymol command file. No use of mechanize.
## If one points to an local .pka file, then method is auto-changed to method=file. This is handsome in off-line environment, ex. teaching or seminar.
# pkafile: Write the path to .pka file. Ex: pkafile=./Results_propka/4ins_.pka
# molecule : name of the molecule. Ending of file is assumed to be .pdb
# chain : which chains are saved to file, before molecule file is send to server. Separate with "." Ex: chain=A.b
# resi : Select by residue number, which residues should be printed to screen and saved to the log file: /Results_propka/_Results.log.
## Separate with "." or make ranges with "-". Ex: resi=35.40-50
# resn : Select by residue name, which residues should be printed to screen and saved to the log file: /Results_propka/_Results.log.
## Separate with "." Ex: resn=cys.tyr
# logtime : Each execution give a set of files with the job id=logtime. If logtime is not provided, the current time is used.
## Normal it usefull to set it empty. Ex: logtime=_
# verbose : Verbose is switch, to turn on messages for the mechanize section. This is handsome to see how mechanize works, and for error searching.
# showresult : Switch, to turn on all results in pymol command window. Ex: showresult=yes
# server_wait=10.0 is default. This defines how long time between asking the server for a result. Set no lower than 3 seconds.
# version=v3.1 is default. This is what version of propka which would be used.
## Possible: 'v3.1','v3.0','v2.0'. If a newer version is available than the current v3.1, a error message is raised to make user update the script.
############################################Input parameters: getpropka############################################
############# The order of input and changable things:
getpropka(PDB="NIL",chain="*",resi="0",resn="NIL",source="upload",PDBID="",logtime=time.strftime("%Y%m%d%H%M%S",time.localtime()),server_wait=3.0,version="v3.1",verbose="no",showresult="no")
# PDB: points the path to a .pdb file. This is auto-set from propka function.
# source : source=upload is default and is set at the propka webpage.
# source=ID, PDBID=4ake , one can print to the command line, the pka value for any official pdb ID. No files are displayed in pymol.
# PDBID: is used as the 4 number/letter pdb code, when invoking source=ID.
Examples
Example 1 - Mutagenesis analysis - part 1
This script was developed with the intention of making analysis of possible mutants easier. For example, the reactivity of Cysteines in FRET maleimide labelling is determined by the fraction of the Cysteine residue which is negatively charged (C-). This fraction is related to its pKa value and the pH of the buffer: f(C-)=1/(10(pK-pH)+1). So, one would be interested in having the lowest possible pKa value as possible. Ideally lower than the pH of the buffer. To analyse where to make the best mutant in your protein, you could do the following for several residues. We do the mutagenesis in the command line, since we then could loop over the residues in the protein.
So, in a loop with defined residues, this could look like the following code. Note, now we are quite happy for the result log file, since it collects the pka for the mutants.
| Download: examples/propka_1.pml | |
| This code has been put under version control in the project Pymol-script-repo | |
reinitialize
import propka
fetch 1ohr, async=0
create 1ohrB3C, 1ohr
hide everything, all
show cartoon, 1ohrB3C
cmd.wizard("mutagenesis")
cmd.do("refresh_wizard")
# To get an overview over the wizard API:
for i in dir(cmd.get_wizard()): print i
# lets mutate chain B residue 3 to CYS. (1ohrB3C)
cmd.get_wizard().set_mode("CYS")
cmd.get_wizard().do_select("/1ohrB3C//B/3")
# Select the first rotamer, which is most probable
cmd.frame(1)
# Apply the mutation
cmd.get_wizard().apply()
# Close wizard
cmd.set_wizard("done")
#OR cmd.wizard(None)
propka resi=3
zoom /1ohrB3C//B/3
Example 2 - Mutagenesis analysis - part 2
To only loop over surface residues, you might want to find these with the script FindSurfaceResidues.
| Download: examples/propka_2.pml | |
| This code has been put under version control in the project Pymol-script-repo | |
reinitialize
fetch 1ohr, async=0
import propka
import surfaceatoms
hide everything, all
### We make it in python blocks, so pymol don't speed ahead.
python
### Se version 2 of script: http://www.pymolwiki.org/index.php/FindSurfaceResidues
# When we import a module in python, the namespace is normally: module.function
resis = surfaceatoms.surfaceatoms(cutoff=10.0)
# We dont wan't to kill the server by sending hundreds of requests. So we select some few.
resis = [resis[10],resis[20],resis[30]]
for resi in resis:
newname="1ohr%s%sC"%(resi[0],resi[1])
cmd.create(newname,"1ohr")
cmd.show("cartoon","1ohr%s%sC"%(resi[0],resi[1]))
cmd.wizard("mutagenesis")
cmd.do("refresh_wizard")
cmd.get_wizard().set_mode("CYS")
selection="/%s//%s/%s"%(newname,resi[0],resi[1])
cmd.get_wizard().do_select(selection)
cmd.frame(1)
cmd.get_wizard().apply()
cmd.set_wizard("done")
# When we import a module in python, the namespace is normally: module.function
# And we see, that propka expect resi to be in "str" format.
# And we don't want the logtime function
propka.propka(resi="%s"%resi[1],logtime="")
selection="/%s//%s/%s"%(newname,resi[0],resi[1])
cmd.select("Mutation%s%s"%(resi[0],resi[1]),"byres %s"%(selection))
print resi
python end
cmd.disable("all")
cmd.enable("1ohr")
cmd.zoom("1ohr")
cmd.show("cartoon","1ohr")
print resis
print("Number of surface mutations: %s"%len(resis))
print("Number of residues in protein: %s"%cmd.count_atoms("1ohr and name CA"))
A little warning though. You need to be carefull about the rotamer you're choosing. It can happen that the first rotamer ends up being in physically non-reasonable contact distance to other residues, so atoms become overlayed. Also, the mutagenesis wizard can have the funny habit of sometimes not adding hydrogens to terminal -C or -N after mutating a residue.
Example 3 - Scan a range of proteins
Could be done with this script
| Download: examples/propka_3.pml | |
| This code has been put under version control in the project Pymol-script-repo | |
reinitialize
import propka
cmd.bg_color("white")
cmd.set("auto_zoom","off")
results = []
python
#resis = [["1DSB","*","30"],["1ERT","*","32"],["2TRX","*","32"],["2TRX","*","35"],["1EGO","*","11"],["1EGO","*","14"],["1MEK","*","36"],["1IUE","*","283"],
#["1PPO","*","25"],["1MEG","*","25"],["1QLP","*","232"]]
resis = [["1DSB","*","30"],["1ERT","*","32"]]
for p,c,r in resis:
cmd.fetch(p,async="0")
cmd.refresh()
pkavalues = propka.propka(molecule=p,chain=c,resi=r,logtime="",makebonds="no")
results.append(pkavalues)
cmd.refresh()
python end
python
for p,c,r in resis:
cmd.enable("%s"%(p))
cmd.show_as("cartoon","%s"%(p))
cmd.select("%s%s"%(p,r),"byres (%s and chain %s and resi %s and resn CYS)"%(p,c,r))
cmd.show("sticks","%s%s"%(p,r))
python end
cmd.zoom("all")
ScriptVersion
Current_Version=20111202
Changelog
- 20111202
- This code has been put under version control. In the project, Pymol-script-repo.
- 20110823
- Fixed some issues with selection algebra of Ligands.
- Now colors Ligands automatically to purple scheme.
- 20110822
- Made the naming scheme consistent, so one can work with multiple proteins, and the grouping still works.
- Bonds to N-terminal and C-terminal did not show up. Fixed.
- If one just write "propka", the "last" molecule in the pymol object list is now assumed, instead of the first. This makes mutagenesis analysis easier.
- The pka difference from assumed standard values are now also displayed. Standard values are set to: pkadictio = {'ASP':3.9, 'GLU':4.3, 'ARG':12.0, 'LYS':10.5, 'HIS':6.0, 'CYS':8.3, 'TYR':10.1}
- The menu size is made bigger, so it can fit the long names for the bonding partners. There's also a slider that you can drag using the mouse
- --it's the tiny tab between the command line and the movie controls. Drag that left or right.
- 20110821
- The resn function was made wrong, only taking the last item of list. Fixed.
- resn= can now also be print to screen/log for ligands like ATP. Ex: resn=CYS.ATP.TRP
- Ligands are now also written to the "stripped-file". "./Results_propka/PDB.stripped"
- Bonds are now also made for Ligands.
- 20110820
- If one points to a result .pka file, then "method" is automatically set to "method=file".
- Added the ability for pka values for ligands
- Bonds are now generated for the pka atoms.
- The color scheme is changed from "rainbow" to "red_white_blue". This is easier to interpret.
- CC: COULOMBIC INTERACTION. Color is red.
- SH: SIDECHAIN HYDROGEN BOND. Color is brightorange.
- BH: BACKBONE HYDROGEN BOND. Color is lightorange.
- 20110817
- If just invoking with "propka", it will select the first molecule. And now it is possible also to write. "propka all".
- Removed the "raise UserWarning" if the script is oudated. Only a warning message is printed.
- 20110816
- Made the execution of the pymol command script silent, by only using cmd.API. This will raise a Warning, which can be ignored.
- Built-in a Script version control, to inform the user if the propka script is updated on this page
- The alternate attribute for the labeling atoms are reset. It was found pymol objected altering names for atoms which had alternate positions/values.
- Reorganized the input order, which means that: molecule=all is default.
Known bugs
- Bonds can be multiplied from amino acids to Ligands like ZN or ATP. Assume the shortest bond to be correct.
- ZN: This is caused, since ZN has no ID number and when there are several in the same chain. This can be reproduced for 1HP1, bond: A41ZNCC. Here it shows to bonds, where only the shortes is correct. No fix possible.
- ATP: This is caused, since propka sometimes eats the ending of the atom name. In the .pdb file is written O3', which propka represents like O3. Therefore a wildcard "*" is generel inserted, which can cause multiple bonds to the ATP molecule. This can be reproduced for 1HP1, bond: A504ATPSH. Here multiple bonds are made to all the O3*,O2* atoms. No fix possible.
- The propka server sometimes use another naming scheme for Ligands. These Ligands will not be pka labelled or shown in pymol. The results will still downloaded to "/.Results_propka/file.pka"
- This can be reproduced with 1BJ6. Here the propka webpage predicts the pka values for the ligands: AN7,GN1,CN3,CO2,GO2, but the .pdb file names these ligands: DA,DA,DC,DG.
- Alternative configurations of a Ligand is at the moment a problem, and will not be shown. For example 1HXB and the ligand ROC.
- The script extraxt AROC and BROC from pymol, which does match with ROC in the .pka file. Try save the protein with only one configuration of the Ligand.
- >In pymol:
- fetch 1hxb, async=0
- create 1hxbA, 1hxb and not alt B
- save 1hxbA.pdb, 1hxbA
- >Quit pymol
- >Manually replace with text editor in .pdb file "AROC" with " ROC" Remember the space " "!
- >Then in pymol
- load 1hxbA.pdb
- import propka
- propka verbose=yes